
Waargenomen bij één op de 100.000 pasgeborenen, is het syndroom van Pfeiffer geassocieerd met de mutatie van de FGFR1- en FGFR2-genen; beide genen hebben de taak om de fusie van de schedelhechtingen en de ontwikkeling van de vingers en tenen te reguleren.
Voor de diagnose van het syndroom van Pfeiffer zijn een lichamelijk onderzoek, anamnese, een radiologisch onderzoek van de schedel en vingers en tenen en tenslotte een genetische test van fundamenteel belang.
Momenteel kunnen mensen die lijden aan het syndroom van Pfeiffer alleen rekenen op symptomatische behandelingen, dat wil zeggen behandelingen die de symptomen verlichten.
Kort overzicht van de schedelhechtingen en hun fusie
De schedelnaden zijn de fibreuze gewrichten, die dienen om de botten van het schedelgewelf (dwz de frontale, temporale, pariëtale en occipitale botten) samen te smelten.
Onder normale omstandigheden vindt het proces van fusie van de schedelhechtingen plaats in de postnatale periode, beginnend op 1-2 jaar, voor sommige gewrichtselementen, en eindigend op de leeftijd van 20, voor anderen. Dit lange en getrapte proces van fusie stelt de hersenen in staat om adequaat te groeien en zich te ontwikkelen.
- De aanwezigheid van abnormaal grote en afwijkende duimen en grote tenen op een zodanige manier dat ze weg lijken te bewegen van de andere tenen (mediale deviatie).
Het Pfeiffer-syndroom is daarom een genetische aandoening die, bij degenen die het drager zijn, voornamelijk afwijkingen in de schedel en handen bepaalt.
Aangezien lezers de kans krijgen om meer te leren in het hoofdstuk dat gewijd is aan symptomen, kan het syndroom van Pfeiffer echter in verband worden gebracht met andere problemen en andere lichamelijke misvormingen.
Epidemiologie: hoe vaak komt het syndroom van Pfeiffer voor?
Volgens statistieken wordt één op de 100.000 mensen geboren met het Pfeiffer-syndroom.
Wist je dat ...
De genetische ziekten die, zoals het syndroom van Pfeiffer, craniosynostose veroorzaken, zijn ongeveer 150.
Hiervan zijn, naast het Pfeiffer-syndroom, het Crouzon-syndroom, het Apert-syndroom en het Saethre-Chotzen-syndroom van belang.
Wat veroorzaakt de genmutatie geassocieerd met het syndroom van Pfeiffer?
Stelling: de genen die aanwezig zijn op menselijke chromosomen zijn DNA-sequenties die tot taak hebben fundamentele eiwitten te produceren in biologische processen die essentieel zijn voor het leven, inclusief celgroei en replicatie.
Wanneer ze vrij zijn van mutaties (dus bij een gezond persoon), produceren de FGFR1- en FGFR2-genen in de juiste hoeveelheden respectievelijk de fibroblastgroeifactorreceptor 1 en de fibroblastgroeifactorreceptor 2, twee receptoreiwitten die essentieel zijn om de timing van craniale hechtingsfusie en om de ontwikkeling van de vingers en tenen te reguleren (met andere woorden, ze geven aan wanneer het de juiste tijd is voor craniale hechtingsfusie en regelen de vorming van de vingers en voeten).
Aan de andere kant, wanneer ze de mutaties ondergaan die worden waargenomen in aanwezigheid van het Pfeiffer-syndroom, zijn de FGFR1- en FGFR2-genen hyperactief en produceren ze de bovengenoemde receptoreiwitten in zulke enorme hoeveelheden, dat de fusietijden van de schedelhechtingen veranderen (ze zijn sneller ) en het trainingsproces van de vingers en tenen verloopt niet correct.
Het syndroom van Pfeiffer is een autosomaal dominante ziekte
Begrijpen...
Elk menselijk gen is aanwezig in twee exemplaren, allelen genaamd, één van moederlijke oorsprong en één van vaderlijke oorsprong.
Het syndroom van Pfeiffer heeft alle kenmerken van een autosomaal dominante ziekte.
Een genetische ziekte is autosomaal dominant wanneer de mutatie van een enkele kopie van het gen dat het veroorzaakt voldoende is om zich te manifesteren.
Soorten Pfeiffer-syndroom
In 1993 publiceerde de Amerikaanse arts Michael Cohen, na talrijke studies over het syndroom van Pfeiffer, een typologische classificatie van de genetische ziekte in kwestie, die het bestaan voorspelde van drie pathologische varianten, eenvoudig geïdentificeerd met de termen "Type I", "Type II" en Type III "en alle delen de aanwezigheid van craniosynostose en afwijkingen van de duim en grote tenen. De medisch-wetenschappelijke gemeenschap accepteerde deze classificatie onmiddellijk en sindsdien gebruiken de experts van het Pfeiffer-syndroom het als een diagnostisch hulpmiddel en voor het beoordelen van de ernst van de aanwezige genetische aandoening; in feite moet worden opgemerkt dat de classificatie van Dr. Cohen het syndroom van Pfeiffer onderscheidt op basis van de ernst van de schedel- en digitale anomalieën en de aanwezigheid van andere symptomen en tekenen.
Als we ingaan op de details van de individuele pathologische varianten, is het op dit punt van het artikel belangrijk om te onderstrepen dat:
- De Typ ik. het is de minder ernstige versie van het syndroom van Pfeiffer, omdat craniostenose en duim- en grote teenafwijkingen beperkte gevolgen hebben.
Andere belangrijke informatie: het is te wijten aan de FGFR2-mutatie, soms gecombineerd met de FGFR1-mutatie; het kan een erfelijke of verworven aandoening zijn. - De Type II het is de meest ernstige versie van het syndroom van Pfeiffer, omdat het gepaard gaat met ernstige craniosynostose, bijna onverenigbaar met het leven, en met ernstige afwijkingen aan handen en voeten.
Andere belangrijke informatie: het is uitsluitend te wijten aan de FGFR2-mutatie; het is altijd een verworven toestand. - De Type III het is de versie van het syndroom van Pfeiffer die, op een schaal van ernst, net onder type II valt, maar ruim boven type I, aangezien de huidige craniosynostose bijna net zo ernstig is als die van de variant die in het vorige punt is beschreven.
Andere belangrijke informatie: het is uitsluitend te wijten aan de FGFR2-mutatie; het is altijd een verworven toestand.
Craniostenose
Bij dragers van het syndroom van Pfeiffer kan craniosynostose, afhankelijk van het aantal schedelnaden dat betrokken is bij het vroege fusieproces, de volgende gevolgen hebben:
- Totaal abnormale verticale ontwikkeling van het hoofd, gecombineerd met een gebrek aan laterale expansie van de schedel. Daarom heeft de patiënt met het Pfeiffer-syndroom een lang, smal hoofd;
- Vorming van een hoog en prominent voorhoofd;

- Verhoogde intracraniale druk, waarvan symptomen zoals aanhoudende hoofdpijn, problemen met het gezichtsvermogen, braken, prikkelbaarheid, gehoorproblemen, ademhalingsproblemen, veranderingen in de mentale toestand, papiloedeem afhankelijk zijn;
- Intellectuele tekorten die leiden tot een verminderd IQ. Intellectuele tekortkomingen zijn het gevolg van de verminderde ruimte voor groei die de hersenen genieten nadat de coronale schedelhechtingen voortijdig zijn samengesmolten;
- Gebrek aan ontwikkeling van het tussenliggende deel van het gezicht, dat vlak, zo niet hol lijkt;
- Aanwezigheid van uitpuilende (proptosis), wijd open en abnormaal uit elkaar geplaatste ogen (oculair hypertelorisme);
- Aanwezigheid van een snavelneus;
- Het niet ontwikkelen van de kaak (maxillaire hypoplasie), resulterend in een toestand van overvolle tanden;
- Klaverachtig uiterlijk van het hoofd ("klaverschedel"). De "klaverschedel" veroorzaakt hydrocephalus.
TYPE I
Type I Pfeiffer-syndroom wordt geassocieerd met een milde klinische craniosynostose, die zich vaak beperkt tot het geven van een langwerpige vorm aan de schedel en het veroorzaken van een zichtbaar hoog voorhoofd en een plat gezicht.
Als ze de juiste behandeling krijgen, leiden mensen met het Pfeiffer-syndroom type I meestal een normaal leven en hebben ze een normaal IQ.
TYPE II
Type II Pfeiffer-syndroom is de enige pathologische variant die de zogenaamde "klaverbladschedel" veroorzaakt, deze schedelafwijking heeft ernstige gevolgen voor intellectuele vermogens en wordt vaak geassocieerd met vroegtijdige dood.
Degenen die lijden aan Type II Pfeiffer-syndroom presenteren het hele klinische beeld dat hierboven is beschreven met betrekking tot de gevolgen van craniosynostose.

TYPE III
Type III Pfeiffer-syndroom heeft dezelfde impact op zijn dragers als Type II Pfeiffer-syndroom, behalve de "klaverbladschedel".
Mensen met Type III Pfeiffer Syndroom hebben geen lange levensverwachting.
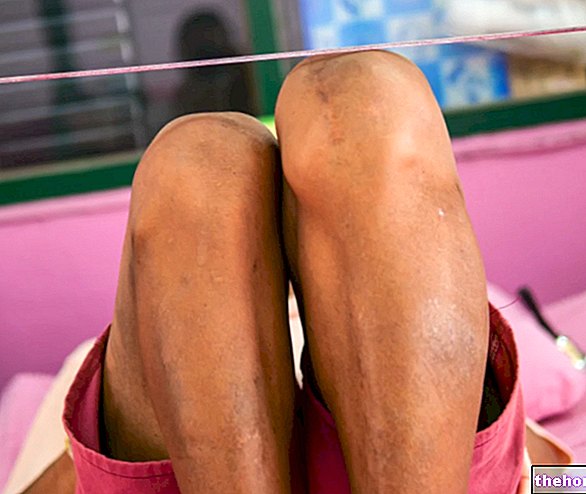
Afwijkingen die de duimen en grote tenen aantasten
Indien bijzonder ernstig, kunnen de afwijkingen die de duimen en grote tenen aantasten de functionele capaciteit van de handen en voeten ernstig aantasten, waardoor problemen ontstaan bij het grijpen van voorwerpen en/of lopen.
Wist je dat ...
De mediale afwijking van de duimen en grote tenen van patiënten met het Pfeiffer-syndroom is een voorbeeld van varus varus. Om precies te zijn spreken artsen van duim varus, vanwege de mediale afwijking van de duimen, en hallux varus, vanwege de mediale afwijking van de grote tenen.

brachydactylie
Bij het syndroom van Pfeiffer is brachydactylie een vrij veel voorkomende afwijking die slechts enkele vingers of het gehele digitale complex van de handen en/of voeten kan aantasten.
Het probleem van brachydactylie is waarneembaar in alle typologische varianten, zij het met een verschillende frequentie.
syndactylie
Bij het syndroom van Pfeiffer vormt syndactylie een vrij frequente "afwijking (minder vaak voor dan brachydactylie), die verschillende connotaties kan hebben (het kan onvolledig, compleet, complex, enz.) zijn.
Het probleem van brachydactylie is waarneembaar in alle typologische versies van het Pfeiffer-syndroom, zij het met verschillende recidieven.
Botankylose
Het syndroom van Pfeiffer wordt vooral geassocieerd met ankylose van de elleboog, hoewel het in werkelijkheid hetzelfde probleem kan veroorzaken bij elk groot gewricht in het menselijk lichaam.
Botankylose is een probleem dat alleen voorkomt bij de meest ernstige typologische versies van het syndroom van Pfeiffer (vooral bij type II).
Afwijkingen die de luchtwegen aantasten
De mogelijke anomalieën in de luchtwegen veroorzaakt door het syndroom van Pfeiffer zijn van dien aard dat ze ademhalingsproblemen veroorzaken met ernstige gevolgen voor de algemene gezondheid van de patiënt (de hersenen lijden het meest).
Net als botankylose zijn de bovengenoemde anomalieën alleen waarneembaar in de meer ernstige typologische varianten (met name Type II).
Wanneer is het mogelijk om het syndroom van Pfeiffer te detecteren?
Doorgaans zijn schedel- en digitale afwijkingen als gevolg van het Pfeiffer-syndroom duidelijk bij de geboorte, dus diagnose en behandelingsplanning zijn onmiddellijk.
aan het hoofd (hoofdröntgenfoto's, hoofd-CT en/of hoofd-MRI) en handen en voeten; ten slotte eindigt het met een genetische test.
Lichamelijk onderzoek en medische geschiedenis
Lichamelijk onderzoek en anamnese bestaan in wezen uit een nauwkeurige evaluatie van de symptomen die de patiënt vertoont.
In de context van het syndroom van Pfeiffer stelt de arts in deze fasen van het diagnostisch proces de craniostenose en de afwijkingen aan duimen en grote tenen vast en stelt hij op basis van de andere aanwezige symptomen een hypothese op over de lopende typologische variant.
Radiologisch onderzoek van hoofd en vingers en tenen
In de context van het syndroom van Pfeiffer,
- Radiologische onderzoeken van het hoofd worden door de arts gebruikt om de aanwezigheid van een vroege fusie van de schedelhechtingen te bevestigen en om de ernst van de schedel-hersenafwijkingen in te schatten.
- Radiologisch onderzoek daarentegen is essentieel om de omvang van de varus en een "mogelijke brachydactylie en/of" een mogelijke syndactylie te onderzoeken.
genetische test
Het is de DNA-analyse gericht op het opsporen van mutaties in kritische genen.
In de context van het Pfeiffer-syndroom vertegenwoordigt het de bevestigende diagnostische test, omdat het de mutatie van FGFR2 en / of FGFR1 kan benadrukken.
De genetische test is ook de test waarmee het type Pfeiffer-syndroom kan worden vastgesteld.




-cos-cause-e-terapia.jpg)



















-cos-cause-e-trattamento.jpg)