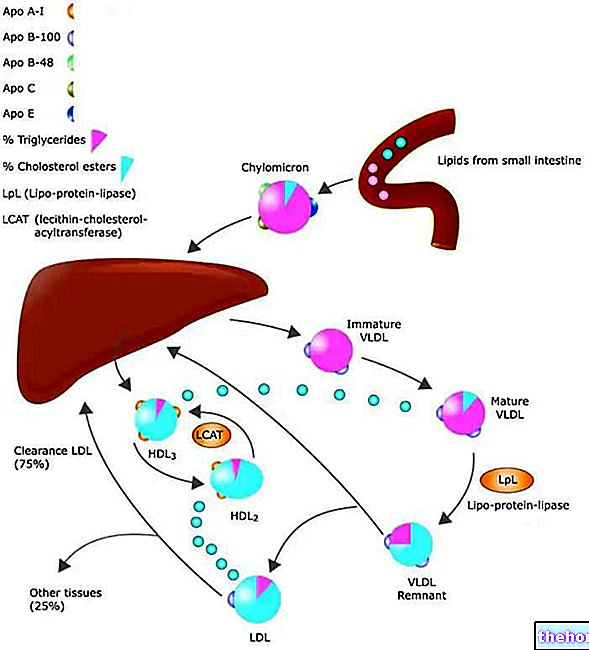
Lipoproteïnelipase (LPL) is een enzym dat wordt aangetroffen in endotheelcellen die het binnenoppervlak van bloedcapillairen bekleden. Het is met name geconcentreerd op het niveau van het capillaire endotheel van skeletspierweefsel, hartweefsel en vetweefsel. Het is niet verrassend dat de functie van het lipoproteïnelipase is om de triglyceriden in de lipoproteïnen (chylomicronen en VLDL) te hydrolyseren, waarbij twee vetzuren vrijkomen. vrij en een monoacylglycerol.De producten die voortkomen uit deze hydrolyse van triglyceriden diffunderen in de cellen, waar ze in wezen twee doelen kunnen bereiken: de eerste moet worden gemetaboliseerd in de skeletspier en in het hart, de tweede, typisch voor de perioden van overvoeding (energieoverschot), moet worden gebruikt als substraat voor de hersynthese van triglyceriden en vervolgens worden geaccumuleerd als energiereserve.
Insuline verhoogt de expressie van lipoproteïnelipase op het niveau van wit vetweefsel, wat de hydrolyse van bloedtriglyceriden in glycerol en vetzuren bevordert; deze laatste kunnen dus de adipocyten binnendringen en vervolgens opnieuw worden veresterd met glycerol, waardoor reservetriglyceriden worden gevormd.
Familiale lipoproteïne-lipasedeficiëntie (ziekte van Burger-Grutz of familiale hyperlipoproteïnemie type I)
Autosomaal recessieve ziekte, met een incidentie gelijk aan één geval op 100.000 mensen. Het komt voor bij personen die homozygoot zijn voor een mutatie op het gen dat codeert voor lipoproteïnelipase. Het daaruit voortvloeiende tekort aan dit enzym zorgt ervoor dat degenen die door deze ziekte zijn getroffen, bijzonder hoge triglyceridenspiegels vertonen (meestal meer dan 800-1000 mg / dL), vanwege de blokkering van het metabolisme van chylomicronen. Ernstige hypertriglyceridemie gaat sinds de kindertijd gepaard met een grotere incidentie van pancreatitis, buikpijn, eruptieve xanthomen (geelachtige papels met rode contouren verdeeld over de lichaamsdelen die onder druk staan) en hepatosplenomegalie (abnormale vergroting van de lever en milt). cardiovasculair risico, terwijl retinopathie soms aanwezig is.
Familiale APO-C2-deficiëntie
Een heel belangrijk eiwit, omdat het in staat is om het lipoproteïne lipase te activeren, is het Apo-lipo-eiwit-C2 of APO-C2. Een tekort aan dit eiwit, uitgedrukt op het oppervlak van VLDL en chylomicronen, kan hyperlipoproteïnemie veroorzaken die wordt gekenmerkt door hypertriglyceridemie (hoge triglyceriden in het bloed).APO-C2-deficiëntie wordt dan ook geassocieerd met een verhoogd risico op vroege atherosclerose en op pancreatitis, wat vaker voorkomt bij ouderen. leeftijd. Ook hier is de ziekte gekoppeld aan een autosomaal recessieve mutatie, namelijk in het gen dat codeert voor APO-C2.
Lipoproteïnelipasen, voeding, medicijnen en supplementen
Lipoproteïnelipase of APO-C2-deficiëntie kan worden behandeld met een vetarm dieet, geconsumeerd in hoeveelheden van niet meer dan 10-20 gram per dag. Vetten met tussenliggende ketens, die direct aan albumine binden en geen gebruik maken van chylomicronen om in de bloedbaan te worden getransporteerd, verdienen duidelijk de voorkeur. Tegelijkertijd moet alcohol worden afgeschaft en moet worden gezorgd voor een adequate toevoer van in vet oplosbare vitamines en vetzuren essentieel. Vooral de omega-3 vetzuren uit vis (EPA en DHA) hebben opmerkelijke hypotiglyceridenverlagende eigenschappen laten zien en worden als zodanig in hoge doses gebruikt om de triglyceridenspiegels te verlagen. Andere geneesmiddelen met vergelijkbare activiteit zijn fibraten en nicotinezuur, die hun hypotryglycerideverlagende werking ook uitoefenen door de expressie van het enzym lipoproteïnelipase te verhogen.