Algemeenheid
De voorwaarde retinitis pigmentosa (RP) identificeert een groep genetische ziekten die worden gekenmerkt door progressieve retinale degeneratie.

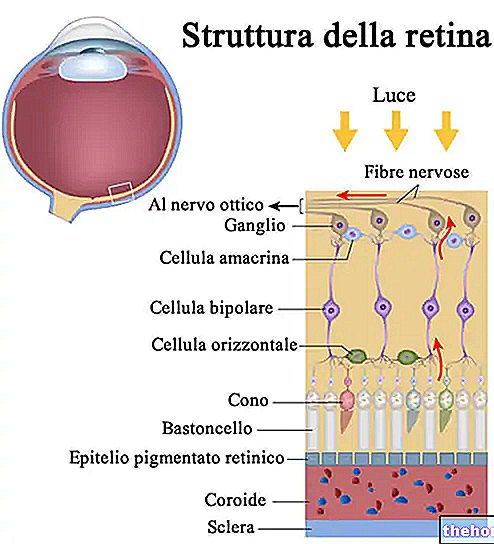
Retinitis pigmentosa is een retinale dystrofie die wordt gekenmerkt door het geleidelijke verlies van fotoreceptoren en disfunctie van het pigmentepitheel, wat betekent dat het netvlies geleidelijk zijn vermogen vermindert om visuele informatie via de oogzenuw naar de hersenen te verzenden.
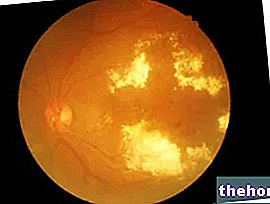
Het pathologische proces begint met veranderingen van het retinale pigmentepitheel. Naarmate retinitis pigmentosa vordert, is er een dunner worden van de bloedvaten die het netvlies voeden, die atrofie ondergaan. Bij onderzoek van de fundus zijn de karakteristieke afzettingen visueel detecteerbaar. retinaal pigment ( vandaar de naam van de ziekte). Atrofische veranderingen en schade kunnen ook betrekking hebben op de oogzenuw en geleidelijk sterven de lichtgevoelige cellen van het netvlies af.
Patiënten met retinitis pigmentosa ervaren aanvankelijk problemen met het gezichtsvermogen, vooral in slecht verlichte omgevingen en klagen over een vernauwing van het perifere gezichtsveld. Het centrale zicht wordt gespaard tot de latere stadia van de ziekte, en het uiteindelijke resultaat kan dramatisch variëren: veel mensen met retinitis pigmentosa behouden hun hele leven een beperkt gezichtsvermogen, terwijl anderen het gezichtsvermogen volledig verliezen.
Retinitis pigmentosa is een erfelijke ziekte die voornamelijk wordt veroorzaakt door genetische veranderingen die door een of beide ouders zijn doorgegeven. Het type genetisch defect bepaalt welke netvliescellen het meest betrokken zijn bij de aandoening en maakt het mogelijk om de verschillende aandoeningen klinisch te onderscheiden. Tot op heden zijn meer dan 50 verschillende genetische defecten geïdentificeerd die betrokken zijn bij retinitis pigmentosa. Afwijkingen kunnen van ouders op nakomelingen worden doorgegeven via een van de drie overervingspatronen: autosomaal recessief, autosomaal dominant of heterosomaal recessief (X-gebonden of X-gebonden).
Symptomen
Voor meer informatie: Retinitis Pigmentosa Symptomen
Retinitis pigmentosa wordt meestal gevonden bij adolescenten en jonge volwassenen. Symptomen treden vaak op tussen de leeftijd van 10 en 30 jaar, maar de diagnose kan in de vroege kinderjaren of veel later in het leven worden gesteld.
Vroege symptomen van retinitis pigmentosa kunnen zijn:
- Moeite met zien 's nachts (nachtblindheid) of bij weinig licht
- Langzame aanpassing van zicht in het donker naar dat in het licht, en vice versa;
- Vernauwing van het gezichtsveld en verlies van perifeer zicht;
- Gevoeligheid voor licht en verblinding.
Sommige symptomen zijn afhankelijk van het type betrokken fotoreceptoren. De staafjes zijn verantwoordelijk voor het zwart-witzicht, terwijl de kegeltjes je in staat stellen om kleuren te onderscheiden.
In de meeste gevallen van retinitis pigmentosa zijn eerst de staafjes betrokken. In de snel evoluerende vormen kunnen kegels echter ook in een vroeg stadium worden aangetast.
Staafjes zijn geconcentreerd in de buitenste delen van het netvlies en worden geactiveerd door zwak licht, zodat hun degeneratie het perifere en nachtzicht beïnvloedt. Als er kegeltjes zijn, is het mogelijk om verlies van kleurwaarneming en centraal zicht te ervaren.
Het overwicht van de betrokken fotoreceptoren wordt bepaald door het specifieke defect dat aanwezig is in de genetische samenstelling van de patiënt.
Vaak is nachtblindheid (of nocthalopie) het eerste symptoom van retinitis pigmentosa. Sommige mensen merken dat ze meer en meer tijd nodig hebben om zich aan te passen aan verschillen in licht als ze van een goed verlichte naar een donkerdere gaan. Een typische vorm van gezichtsverlies veroorzaakt vernauwing van het perifere zicht (tunnel- of telescoopzicht); dit patroon wordt een ringscotoma genoemd. Soms ontbreekt dit fenomeen in de vroege stadia, maar het wordt opgemerkt wanneer het individu vaak over objecten struikelt of betrokken is bij een verkeersongeval.Wanneer gezichtsverlies het centrale gebied van het netvlies betreft (ook wel maculaire dystrofie genoemd) moeite hebben met lezen en gedetailleerd werk dat concentratie op een enkel object vereist, zoals een draad door het oog van een naald halen Veel patiënten melden dat ze lichtflitsen (fotopsie) zien, vaak beschreven als kleine, flikkerende en twinkelende lichtjes.
De snelheid van ziekteprogressie en de mate van gezichtsverlies variëren van persoon tot persoon. Sommige extreme gevallen kunnen zich snel ontwikkelen binnen twee decennia, andere een langzaam verloop dat nooit tot volledige blindheid leidt. Vroege aanvang wordt gevonden bij ernstigere vormen van retinitis pigmentosa, terwijl patiënten met mildere aandoeningen (bijv. autosomaal dominant) de ziekte in hun vijfde of zesde levensdecennium kunnen ontwikkelen.In families met X-gebonden retinitis pigmentosa worden mannen vaker getroffen dan vrouwen en ernstiger; vrouwen daarentegen dragen de genetische eigenschap over (zij dragen het gewijzigde gen op het X-chromosoom) en vertonen minder vaak symptomen van de aandoening.
Complicaties
Retinitis pigmentosa zal blijven vorderen, zij het langzaam. Volledige blindheid is echter zeldzaam, maar er kan een significante vermindering van het perifere en centrale zicht optreden.
Patiënten met retinitis pigmentosa ontwikkelen vaak op jonge leeftijd zwelling van het netvlies (maculair oedeem) of cataract. Deze complicaties kunnen worden behandeld als ze het gezichtsvermogen verstoren.
Verwante ziekten
Gewoonlijk heeft een patiënt met retinitis pigmentosa geen andere aandoeningen en in dit geval spreken we van "niet-syndromale" of eenvoudige retinitis pigmentosa. Verschillende syndromen delen echter enkele klinische symptomen met deze oogziekte; de meest voorkomende is het syndroom van Usher, dat ongeveer 10-30% van alle patiënten met retinitis pigmentosa treft en gepaard gaat met gelijktijdig aangeboren of progressief gehoorverlies. Bij aangeboren amaurose van Leber kunnen kinderen echter binnen de eerste zes maanden van hun leven blind of bijna blind worden.Andere ziekten die verband houden met retinitis pigmentosa zijn het syndroom van Bardet-Biedl en de ziekte van Refsum.
Oorzaken
De ziekte kan worden veroorzaakt door een aantal genetische defecten: in feite zijn er verschillende genen die, indien aangetast door de wijziging, het fenotype retinitis pigmentosa kunnen veroorzaken.Deze coderen normaal gesproken voor eiwitten die betrokken zijn bij de transductiecascade die visie mogelijk maakt, factoren celtranscriptie (die foutieve berichten naar retinale cellen sturen) of voor elementen die de structuur van fotoreceptoren vormen Erfelijke genmutaties zijn aanwezig in cellen vanaf het moment van conceptie; veel voorkomende afwijkingen zijn die van RP1-genen (bij retinitis pigmentosa-1, autosomaal dominant) , RHO (RP4, autosomaal dominant) en RDS (RP7, autosomaal dominant) Niet-erfelijke oorzaken van retinitis pigmentosa zijn zeldzaam, maar de mogelijkheid om een geïsoleerd geval te vinden (spontane mutatie), waarin het niet aanwezig is een familiegeschiedenis van de ziekte.







.jpg)