Actieve ingrediënten: Adalimumab
Humira 40 mg oplossing voor injectie in voorgevulde pen
Humira bijsluiters zijn beschikbaar voor verpakkingsgrootten:- Humira 40 mg / 0,8 ml oplossing voor injectie voor pediatrisch gebruik
- Humira 40 mg oplossing voor injectie in voorgevulde spuit
- Humira 40 mg oplossing voor injectie in voorgevulde spuit met naaldbeveiliging
- Humira 40 mg oplossing voor injectie in voorgevulde pen
Waarom wordt Humira gebruikt? Waar is het voor?
Humira bevat de werkzame stof adalimumab, een selectief immuunonderdrukkend middel. Humira is geïndiceerd voor de behandeling van reumatoïde artritis, polyarticulaire juveniele idiopathische artritis, enthesitis-geassocieerde artritis, spondylitis ankylopoetica, axiale spondylartritis zonder radiografisch bewijs van spondylitis ankylopoetica, artritis psoriatica, psoriatische ettering, de ziekte van Crohn en colitis ulcerosa. het ontstekingsproces van deze ziekten De werkzame stof, adalimumab, is een humaan monoklonaal antilichaam geproduceerd door celculturen Monoklonale antilichamen zijn eiwitten die andere eiwitten herkennen en eraan binden Adalimumab bindt aan een specifiek eiwit (tumornecrosefactor of TNFα) dat in hoge concentraties aanwezig bij ontstekingsziekten zoals reumatoïde artritis, polyarticulaire juveniele idiopathische artritis, enthesitis-geassocieerde artritis, spondylitis ankylopoetica, axiale spondylartritis zonder radiografisch bewijs van spondylitis ankylopoetica, artritis psoriatica, psoriasis, hidradenitis suppurativa, de ziekte van Crohn en colitis ulcerosa.
Reumatoïde artritis
Reumatoïde artritis is een ontstekingsziekte van de gewrichten.
Humira wordt gebruikt voor de behandeling van reumatoïde artritis bij volwassenen.Als u matige tot ernstige reumatoïde artritis heeft, kunnen in eerste instantie andere ziektemodificerende geneesmiddelen zoals methotrexaat worden gebruikt. Als de respons op deze medicijnen niet bevredigend is, krijgt u Humira voor de behandeling van reumatoïde artritis.
Humira kan ook worden gebruikt voor de behandeling van ernstige, actieve en progressieve reumatoïde artritis zonder voorafgaande behandeling met methotrexaat.
Van Humira is aangetoond dat het de progressie van schade aan het kraakbeen en bot in de gewrichten, veroorzaakt door de ziekte, vertraagt en het fysieke functioneren verbetert.
Humira wordt over het algemeen gebruikt in combinatie met methotrexaat. Als uw arts besluit dat behandeling met methotrexaat niet geschikt is, kan Humira alleen worden gegeven.
Polyarticulaire juveniele idiopathische artritis en enthesitis-geassocieerde artritis
Polyarticulaire juveniele idiopathische artritis en enthesitis-geassocieerde artritis zijn ontstekingsziekten.
Humira wordt gebruikt voor de behandeling van polyarticulaire juveniele idiopathische artritis bij kinderen en adolescenten van 2 tot 17 jaar en enthesitis-geassocieerde artritis bij kinderen en adolescenten van 6-17 jaar. Andere ziektemodificerende geneesmiddelen, zoals methotrexaat, kunnen bij de diagnose worden gegeven. Als de respons op deze geneesmiddelen niet adequaat is, krijgt u Humira voor de behandeling van polyarticulaire juveniele idiopathische artritis of enthesitis-geassocieerde artritis.
Spondylitis ankylopoetica en axiale spondyloartritis zonder röntgenologisch bewijs van spondylitis ankylopoetica
Spondylitis ankylopoetica en axiale spondyloartritis zonder röntgenologisch bewijs van spondylitis ankylopoetica zijn ontstekingen van de wervelkolom.
Humira wordt gebruikt voor de behandeling van spondylitis ankylopoetica en axiale spondylartritis zonder röntgenologisch bewijs van spondylitis ankylopoetica bij volwassenen. Als u spondylitis ankylopoetica of axiale spondyloartritis heeft zonder röntgenologisch bewijs van spondylitis ankylopoetica, zult u eerst andere medicijnen gebruiken. Als u met deze geneesmiddelen niet adequaat kunt reageren, zult u Humira gebruiken om de tekenen en symptomen van de ziekte te verminderen.
Psoriatische arthritis
Artritis psoriatica is een ontsteking van de gewrichten die gepaard gaat met psoriasis Humira wordt gebruikt voor de behandeling van artritis psoriatica bij volwassenen.
Van Humira is aangetoond dat het de door de ziekte veroorzaakte schade aan kraakbeen en bot in de gewrichten vertraagt en het lichamelijk functioneren verbetert.
Plaque psoriasis bij volwassenen en kinderen
Plaque psoriasis is een huidaandoening die roodachtige, schilferige, verharde plekken op de huid veroorzaakt die bedekt zijn met zilverachtige schubben. Psoriasis wordt vermoedelijk veroorzaakt door een probleem met het immuunsysteem van het lichaam dat leidt tot een verhoogde productie van huidcellen.
Humira wordt gebruikt voor de behandeling van matige tot ernstige plaque psoriasis bij volwassenen. Als u een volwassene bent met matige tot ernstige plaque psoriasis, zult u eerst andere medicijnen gebruiken of fototherapie ondergaan. Als u geen bevredigende respons op deze behandelingen ontwikkelt, krijgt u Humira om de tekenen en symptomen van psoriasis te verminderen.
Humira wordt ook gebruikt voor de behandeling van ernstige plaque psoriasis bij kinderen en adolescenten van 4 tot 17 jaar bij wie lokale therapie en fototherapie niet optimaal hebben gewerkt of niet geïndiceerd zijn.
Suppuratieve hydradenitis
Hidradenitis suppurativa (ook wel acne inversa genoemd) is een chronische inflammatoire huidziekte en is vaak pijnlijk Symptomen kunnen zijn pijnlijke knobbels en abcessen (cysten) die pus kunnen afvoeren Meestal treft het specifieke delen van de huid, zoals de regio oksels , oksels, binnenkant van de dijen, liezen en billen. Er kunnen zich ook littekens vormen in de getroffen gebieden.
Humira wordt gebruikt voor de behandeling van hidradenitis suppurativa bij volwassenen.Humira kan het aantal knobbels en abcessen dat u heeft, en de pijn die vaak met deze ziekte gepaard gaat, verminderen.
Ziekte van Crohn bij volwassenen en kinderen
De ziekte van Crohn is een "ontsteking van het spijsverteringskanaal.
Humira wordt gebruikt voor de behandeling van de ziekte van Crohn bij volwassenen en kinderen van 6 tot 17 jaar. Als u de ziekte van Crohn heeft, krijgt u eerst andere geneesmiddelen. Als u niet voldoende op deze geneesmiddelen reageert, krijgt u Humira om de typische symptomen van de ziekte van Crohn te verminderen.
Colitis ulcerosa
Colitis ulcerosa is een "ontsteking van de darm".
Humira wordt gebruikt voor de behandeling van colitis ulcerosa bij volwassenen. Als u colitis ulcerosa heeft, neemt u eerst andere medicijnen. Als u met deze geneesmiddelen niet adequaat kunt reageren, zult u Humira gebruiken om de tekenen en symptomen van de ziekte te verminderen.
Contra-indicaties Wanneer Humira niet mag worden gebruikt
Gebruik Humira . niet
- Als u allergisch bent voor adalimumab of voor één van de andere bestanddelen van dit geneesmiddel (vermeld in rubriek 6).
- Als u een "ernstige infectie heeft, waaronder actieve tuberculose (zie "Waarschuwingen en voorzorgen"). Het is belangrijk om uw arts te vertellen als u tekenen of symptomen van een infectie heeft, zoals koorts, wonden, vermoeid gevoel, gebitsproblemen.
- In aanwezigheid van matig of ernstig hartfalen. Het is belangrijk om uw arts te vertellen als er sprake is of is van een ernstige hartaandoening (zie "Wanneer moet u extra voorzichtig zijn met dit middel?').
Voorzorgen bij gebruik Wat u moet weten voordat u Humira inneemt
Praat met uw arts of apotheker voordat u Humira . gebruikt
- Als u allergische reacties krijgt met symptomen zoals beklemd gevoel op de borst, piepende ademhaling, duizeligheid, zwelling of uitslag, stop dan met het innemen van Humira en neem onmiddellijk contact op met uw arts.
- Als u een infectie heeft, waaronder langdurige of plaatselijke infecties (bijvoorbeeld beenulcera), raadpleeg dan uw arts voordat u met Humira begint.Als u twijfelt, neem dan contact op met uw arts.
- U kunt gemakkelijker infecties krijgen terwijl u met Humira wordt behandeld. Dit risico kan toenemen als uw longfunctie wordt aangetast. Deze infecties kunnen ernstig zijn en omvatten tuberculose, infecties veroorzaakt door virussen, schimmels, parasieten of bacteriën, of andere opportunistische infecties en sepsis die in zeldzame gevallen levensbedreigend kunnen zijn. Het is belangrijk om uw arts te informeren over symptomen zoals koorts, wonden, vermoeidheid of gebitsproblemen. Uw arts kan u aanraden tijdelijk te stoppen met Humira.
- Aangezien er gevallen van tuberculose zijn geweest bij patiënten die Humira kregen, moet uw arts controleren of u typische tekenen of symptomen van tuberculose heeft voordat de behandeling met Humira wordt gestart. Dit omvat het verzamelen van een gedetailleerde medische evaluatie die uw medische geschiedenis en geschikte klinische tests omvat (bijvoorbeeld een thoraxfoto en tuberculinetest). De prestaties en resultaten van deze tests moeten worden geregistreerd in de Patiëntenwaarschuwing. Het is erg belangrijk om de arts te vertellen als u ooit tuberculose heeft gehad of als u nauw contact heeft gehad met tuberculosepatiënten. Tuberculose kan optreden tijdens de behandeling, ondanks het feit dat u bent preventief behandeld voor tuberculose Neem onmiddellijk contact op met uw arts als tijdens of na de behandeling symptomen van tuberculose (aanhoudende hoest, gewichtsverlies, lusteloosheid, matige koorts) of andere infecties optreden.
- Vertel het uw arts als u woont of reist naar regio's waar schimmelinfecties, zoals histoplasmose, coccidioidomycose of blastomycose, endemisch zijn.
- Vertel het uw arts als u ooit terugkerende infecties heeft gehad of als u aandoeningen heeft die het risico op infectie verhogen.
- Vertel het uw arts als u drager bent van het hepatitis B-virus (HBV), als u een actieve infectie met het hepatitis B-virus heeft of als u denkt een risico te lopen om het hepatitis B-virus op te lopen. Inname van Humira kan ervoor zorgen dat het hepatitis B-virus opnieuw geactiveerd wordt bij mensen die drager zijn van dit virus. In enkele zeldzame gevallen, vooral als de patiënt wordt behandeld met andere geneesmiddelen die het immuunsysteem onderdrukken, kan reactivering van het hepatitis B-virus levensbedreigend zijn.
- Als u ouder bent dan 65 jaar, kunt u vatbaarder zijn voor infecties tijdens het gebruik van Humira. U en uw arts moeten speciale aandacht besteden aan tekenen van infectie tijdens de behandeling met Humira. Het is belangrijk om uw arts te informeren als symptomen van infecties zoals koorts, wonden , vermoeid gevoel of gebitsproblemen.
- Vertel uw arts vóór een operatie of tandheelkundige ingreep dat u Humira gebruikt. Uw arts kan tijdelijke schorsing aanbevelen.
- Als u demyeliniserende ziekten heeft, zoals multiple sclerose, zal uw arts beslissen of u met Humira moet beginnen.
- Bepaalde vaccins kunnen infecties veroorzaken en mogen niet worden gegeven tijdens de behandeling met Humira. Raadpleeg uw arts voordat u zich laat vaccineren. Bij kinderen wordt aanbevolen om, indien mogelijk, het geplande vaccinatieschema uit te voeren, in overeenstemming met de huidige vaccinatierichtlijnen, voordat de behandeling met Humira wordt gestart. Als u Humira tijdens uw zwangerschap heeft gebruikt, kan uw baby een verhoogd risico hebben om deze infectie te krijgen tot ongeveer 5 maanden na de laatste dosis die u tijdens de zwangerschap heeft ingenomen.Het is belangrijk dat u uw kinderarts of andere beroepsbeoefenaar in de gezondheidszorg vertelt. tijdens de zwangerschap, zodat zij kunnen beslissen wanneer uw baby een vaccinatie moet krijgen.
- In geval van licht hartfalen en gelijktijdige behandeling met Humira, zal uw arts de status van uw hart zorgvuldig moeten evalueren en controleren. Het is belangrijk om uw arts te vertellen over eventuele hartproblemen, zowel in het verleden als in het heden. Als er nieuwe symptomen van hartfalen optreden of als bestaande symptomen verergeren (bijvoorbeeld kortademigheid of zwelling van de voeten), neem dan onmiddellijk contact op met uw arts. Uw arts zal beslissen of u Humira kunt gebruiken.
- Bij sommige patiënten is het lichaam mogelijk niet in staat voldoende bloedcellen aan te maken om infecties te helpen bestrijden of bloedingen te stoppen. Als u aanhoudende koorts, blauwe plekken of gemakkelijk bloeden of bleekheid heeft, neem dan onmiddellijk contact op met uw arts. Deze laatste kan besluiten de therapie te staken.
- Sommige vormen van kanker zijn zeer zelden voorgekomen bij patiënten, zowel kinderen als volwassenen, die werden behandeld met Humira of andere anti-TNF-geneesmiddelen. Patiënten met langdurige ernstige reumatoïde artritis kunnen een hoger dan gemiddeld risico hebben op het ontwikkelen van lymfoom (een type kanker dat het lymfestelsel aantast) en leukemie (een type kanker dat het bloed en het beenmerg aantast). Als u Humira gebruikt, kan het risico op het krijgen van lymfoom, leukemie of andere vormen van kanker toenemen. In zeldzame gevallen is een specifiek en ernstig type lymfoom waargenomen bij patiënten die Humira kregen. Sommige van deze patiënten kregen ook een behandeling met azathioprine of 6-mercaptopurine. Vertel het uw arts als u samen met Humira azathioprine of 6-mercaptopurine gebruikt. Daarnaast zijn gevallen van niet-melanotische huidkanker waargenomen bij patiënten die Humira gebruikten. Als er nieuwe huidlaesies optreden tijdens of na de therapie, of als het uiterlijk van bestaande laesies verandert, vertel dit dan aan uw arts.
- Er zijn gevallen geweest van maligniteiten, naast lymfoom, bij patiënten met een specifiek type longziekte, chronische obstructieve longziekte (COPD) genaamd, die werden behandeld met een ander anti-TNF. Als u COPD heeft of veel rookt, moet u met uw arts bespreken of behandeling met een TNF-blokker geschikt is.
Kinderen en adolescenten
- Vaccinaties: Indien mogelijk moeten kinderen alle vaccinaties hebben gehad voordat ze Humira gebruiken.
- Geef Humira niet aan kinderen met polyarticulaire juveniele idiopathische artritis jonger dan 2 jaar.
Interacties Welke medicijnen of voedingsmiddelen kunnen het effect van Humira . veranderen?
Andere medicijnen en Humira
Vertel het uw arts of apotheker als u andere geneesmiddelen gebruikt, kort geleden heeft gebruikt of in de nabije toekomst gaat gebruiken.
Humira kan worden ingenomen met methotrexaat of andere ziektemodificerende antireumatische geneesmiddelen (sulfasalazine, hydroxychloroquine, leflunomide en parenterale goudzouten), steroïden of analgetica, waaronder niet-steroïde anti-inflammatoire geneesmiddelen (NSAID's).
Humira mag niet gelijktijdig worden ingenomen met geneesmiddelen die anakinra of abatacept als het werkzame bestanddeel bevatten. Als u het niet zeker weet, vraag het dan aan uw arts.
Waarop moet u letten met eten en drinken?
Omdat Humira onder de huid (subcutaan) wordt geïnjecteerd, hebben voedsel en drank geen invloed op Humira.
Waarschuwingen Het is belangrijk om te weten dat:
Zwangerschap en borstvoeding
De effecten van Humira bij zwangere vrouwen zijn niet bekend, daarom wordt het gebruik van Humira bij zwangere vrouwen niet aanbevolen.Het wordt aangeraden om zwangerschap te voorkomen door adequate anticonceptie te gebruiken tijdens de behandeling met Humira en gedurende ten minste 5 maanden na de laatste medicamenteuze behandeling. Als u zwanger wordt, dient u uw arts te raadplegen.
Het is niet bekend of adalimumab overgaat in de moedermelk.
Als u borstvoeding geeft, moet u stoppen met het geven van borstvoeding tijdens de behandeling met Humira en gedurende ten minste 5 maanden na uw laatste behandeling met Humira. Als u Humira tijdens de zwangerschap heeft gebruikt, kan uw baby een verhoogd risico hebben op het krijgen van een infectie.Het is belangrijk dat u uw kinderarts of andere beroepsbeoefenaar in de gezondheidszorg vertelt over uw gebruik van Humira tijdens de zwangerschap, voordat uw baby enige vorm van medicatie krijgt. (zie voor meer informatie de rubriek over vaccinatie).
Als u vermoedt of van plan bent zwanger te worden, vraag dan uw arts of apotheker om advies voordat u dit geneesmiddel gebruikt.
Rijvaardigheid en het gebruik van machines
Humira kan uw vermogen om te rijden, fietsen of machines te bedienen beïnvloeden, al is het maar op een bescheiden manier. Nadat u Humira heeft ingenomen, kunt u gezichtsstoornissen krijgen en het gevoel hebben dat uw omgeving tolt.
Dosering en wijze van gebruik Hoe gebruikt u Humira: Dosering
Gebruik dit geneesmiddel altijd precies zoals uw arts of apotheker u dat heeft verteld. Raadpleeg bij twijfel uw arts of apotheker
Volwassenen met reumatoïde artritis, artritis psoriatica, spondylitis ankylopoetica of axiale spondylartritis zonder röntgenologisch bewijs van spondylitis ankylopoetica.
Humira wordt onder de huid geïnjecteerd (subcutaan gebruik). De gebruikelijke dosering bij volwassen patiënten met reumatoïde artritis, spondylitis ankylopoetica, axiale spondylartritis zonder radiografisch bewijs van spondylitis ankylopoetica en artritis psoriatica is 40 mg adalimumab eenmaal per twee weken, gegeven als een enkele dosis.
Bij reumatoïde artritis wordt methotrexaat voortgezet tijdens de behandeling met Humira.Als uw arts besluit dat methotrexaat niet geschikt is, kan Humira alleen worden gegeven.
Als u reumatoïde artritis heeft en geen methotrexaat krijgt in combinatie met een behandeling met Humira, kan uw arts besluiten om wekelijks 40 mg adalimumab voor te schrijven.
Kinderen met polyarticulaire juveniele idiopathische artritis
De aanbevolen dosis Humira voor patiënten met polyarticulaire juveniele idiopathische artritis in de leeftijd van 2 tot 12 jaar hangt af van de lengte en het gewicht van het kind.De arts van uw kind zal u adviseren over de juiste dosis die u moet gebruiken.
De aanbevolen dosis Humira voor patiënten met polyarticulaire juveniele idiopathische artritis in de leeftijd van 13-17 jaar is 40 mg eenmaal per twee weken.
Kinderen met artritis geassocieerd met enthesitis
De aanbevolen dosis Humira voor patiënten met enthesitis-geassocieerde artritis in de leeftijd van 6 tot 17 jaar hangt af van de lengte en het gewicht van het kind.
Volwassenen met psoriasis
De gebruikelijke dosis Humira voor volwassenen met psoriasis is een startdosis van 80 mg, gevolgd door een dosis van 40 mg, eenmaal per twee weken, te beginnen in de week na de aanvangsdosis. U moet doorgaan met de behandeling met Humira zolang als uw dokter vertelt het je.
Kinderen of adolescenten met plaque psoriasis
De aanbevolen dosis Humira voor patiënten van 4 tot 17 jaar met plaque psoriasis hangt af van het gewicht van het kind. De arts van uw kind zal u vertellen welke dosis u moet gebruiken. Patiënten die een dosis van minder dan 40 mg nodig hebben, moeten Humira gebruiken in de vorm van een injectieflacon van 40 mg.
Volwassenen met hidradenitis suppurativa
De gebruikelijke dosering voor hidradenitis suppurativa is een startdosering van 160 mg (4 injecties op één dag of 2 injecties per dag gedurende twee opeenvolgende dagen), gevolgd door een dosis van 80 mg (2 injecties op dezelfde dag) twee weken later. nog twee weken, ga verder met een dosis van 40 mg per week. Aanbevolen wordt om dagelijks een antiseptische wasoplossing op de aangetaste plekken te gebruiken.
Kinderen of adolescenten met de ziekte van Crohn
Kinderen of adolescenten die minder dan 40 kg wegen:
Het gebruikelijke doseringsschema is 40 mg aan het begin, gevolgd door 20 mg twee weken later.Als een snellere respons nodig is, kan de arts een startdosis van 80 mg voorschrijven (als twee injecties op één dag), gevolgd door 40 mg twee weken later .
Daarna is de gebruikelijke dosering 20 mg eenmaal per twee weken. Afhankelijk van de reactie van het kind kan de arts de frequentie van de dosis verhogen tot 20 mg per week.
Kinderen of adolescenten die 40 kg of meer wegen:
Het gebruikelijke doseringsschema is 80 mg aan het begin, gevolgd door 40 mg twee weken later.Als een snellere respons nodig is, kan de arts een startdosis van 160 mg voorschrijven (als 4 injecties per dag of als 2 injecties per dag gedurende 2 opeenvolgende dagen) gevolgd door 80 mg twee weken later.
Daarna is de gebruikelijke dosering 40 mg eenmaal per twee weken. Afhankelijk van de reactie van het kind kan de arts de doseringsfrequentie verhogen tot wekelijks 40 mg.
Patiënten die een dosis van minder dan 40 mg nodig hebben, moeten Humira gebruiken in de vorm van een injectieflacon van 40 mg.
Volwassenen met colitis ulcerosa
De gebruikelijke dosis Humira voor volwassenen met colitis ulcerosa is 160 mg in week 0 (de dosis kan worden gegeven als vier injecties op één dag of als twee injecties per dag gedurende twee opeenvolgende dagen) en is gelijk aan 80 mg per week. daarna bij 40 mg eenmaal per twee weken. Afhankelijk van de klinische respons kan uw arts de dosis verhogen tot wekelijks 40 mg.
Wijze van toediening en wijze van toediening
Humira wordt toegediend via een injectie onder de huid (via subcutane injectie).
Instructies voor het bereiden en injecteren van Humira:
De onderstaande instructies laten zien hoe u Humira moet injecteren. Lees de instructies aandachtig en volg ze stap voor stap. U krijgt instructies van uw arts of zijn assistent over de techniek van zelftoediening.Injecteer uzelf pas als u zeker weet dat u begrijpt hoe u de toediening moet voorbereiden en toedienen. Na de juiste instructies
De inhoud van de spuit mag niet worden gemengd met andere geneesmiddelen in dezelfde spuit of injectieflacon.
1) Voorbereiding
- Was uw handen grondig.
- Plaats de volgende items op een schoon oppervlak: een voorgevulde spuit Humira voor injectie en een alcoholdoekje.
- Controleer de vervaldatum op de spuit. Gebruik het product niet na de aangegeven maand en het jaar.
2) Keuze en voorbereiding van een injectieplaats
- Kies een plek op de dij of buik.
- Elke nieuwe injectie moet ten minste 3 cm van de plaats van de laatste injectie worden gegeven.Injecteer niet in gebieden waar de huid rood, gekneusd of hard is. Dit kan duiden op een infectie. Veeg de injectieplaats schoon met het alcoholdoekje met een draaiende beweging. Raak het gebied niet meer aan voordat u gaat injecteren.
3) Injectie van Humira
- Schud de spuit NIET.
- Verwijder de dop van de naald van de spuit en zorg ervoor dat u de naald niet aanraakt of dat de naald geen enkel oppervlak raakt.
- Pak met één hand het gebied dat al met alcohol is ingewreven voorzichtig vast en houd het stil.
- Houd met uw andere hand de spuit (met de geribbelde kant naar boven) in een hoek van 45° ten opzichte van de injectieplaats.
- Duw met een stevige, snelle beweging de hele naald in de huid.
- Verlaat de huid met de eerste hand.
- Druk op de zuiger om de oplossing te injecteren - het kan 2 tot 5 seconden duren om de spuit te legen.
- Als de spuit leeg is, haalt u de naald van uw huid en houdt u deze in dezelfde hoek als toen deze werd ingebracht.
- Druk met uw duim of een gaasje gedurende 10 seconden op de injectieplaats. Er kan een kleine bloeding optreden. Masseer de injectieplaats niet. Breng desgewenst een pleister aan.
Verwijdering van materialen
- Gebruik de Humira-spuit NOOIT opnieuw. Sluit de naald NOOIT opnieuw af.
- Gooi na het injecteren van Humira de gebruikte spuit onmiddellijk weg in een speciale container zoals voorgeschreven door uw arts, verpleegkundige of apotheker.
- Houd de container buiten het zicht en bereik van kinderen.
Overdosering Wat moet u doen als u te veel Humira heeft ingenomen?
Wat u moet doen als u meer van Humira heeft gebruikt dan u zou mogen:
Als u Humira per ongeluk vaker injecteert dan is voorgeschreven door uw arts of apotheker, neem dan contact op met uw arts of apotheker om hen te informeren dat u meer geneesmiddel heeft ingenomen. Bewaar de medicijndoos altijd, ook als deze leeg is.
Wat u moet doen wanneer u bent vergeten Humira te gebruiken:
Als u bent vergeten een injectie te geven, moet u uw volgende dosis Humira injecteren zodra u eraan denkt.Vervolg uw dosis dan regelmatig volgens uw normale schema.
Als u stopt met het innemen van Humira
De beslissing om te stoppen met het gebruik van Humira dient met uw arts te worden besproken.De symptomen kunnen terugkeren na stopzetting.
Als u nog vragen heeft over het gebruik van dit geneesmiddel, neem dan contact op met uw arts of apotheker.
Bijwerkingen Wat zijn de bijwerkingen van Humira
Zoals elk geneesmiddel kan ook dit geneesmiddel bijwerkingen hebben, al krijgt niet iedereen daarmee te maken. De meeste bijwerkingen zijn licht tot matig. Sommige kunnen echter ernstig zijn en behandeling vereisen. Bijwerkingen kunnen optreden tot 4 maanden na de laatste Humira-injectie.
Vertel het uw arts onmiddellijk als u een van de volgende reacties opmerkt:
- ernstige huiduitslag, netelroos of andere tekenen van een allergische reactie;
- zwelling van het gezicht, handen, voeten;
- moeite met ademhalen, moeite met slikken;
- kortademigheid bij inspanning of bij het liggen of gezwollen voeten.
Vertel het uw arts zo snel mogelijk als u een van de volgende reacties opmerkt:
- tekenen van infectie zoals koorts, onwel voelen, wonden, gebitsproblemen, branderig gevoel bij het plassen;
- vermoeidheid of zwakte;
- hoest;
- tintelingen;
- doof gevoel;
- dubbel zicht;
- zwakte van de armen of benen;
- zwelling of open zweer die niet geneest
- tekenen en symptomen die wijzen op het optreden van stoornissen die het hematopoëtische systeem beïnvloeden, zoals de aanwezigheid van aanhoudende koorts, blauwe plekken, bloedingen, bleekheid.
De hierboven beschreven symptomen kunnen tekenen zijn van de volgende bijwerkingen die zijn waargenomen bij Humira:
Zeer vaak (kan voorkomen bij meer dan 1 op de 10 mensen):
- reacties op de injectieplaats (waaronder pijn, zwelling, roodheid of jeuk);
- luchtweginfecties (waaronder verkoudheid, rinorroe, sinusitis en longontsteking);
- hoofdpijn;
- buikpijn;
- misselijkheid en braken;
- uitslag;
- musculoskeletale pijn.
Vaak (komen voor bij minder dan 1 op de 10 gebruikers):
- ernstige infecties (waaronder bloedvergiftiging en griep);
- huidinfecties (inclusief cellulitis en herpes zoster-infectie);
- Oor infecties;
- orale infecties (inclusief tandinfecties en herpes simplex);
- infecties van het voortplantingssysteem;
- urineweginfecties;
- schimmelinfecties;
- gewrichtsinfecties;
- goedaardige tumoren;
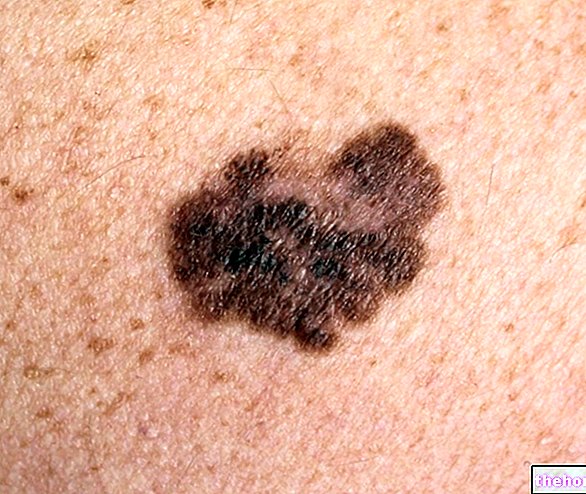
- huidkanker;
- allergische reacties (inclusief seizoensgebonden allergie);
- uitdroging;
- stemmingswisselingen (inclusief depressie);
- ongerustheid;
- slaapproblemen;
- gevoeligheidsstoornissen zoals tintelingen, spiertrekkingen of gevoelloosheid;
- migraine;
- zenuwwortelcompressie (inclusief lage rugpijn en pijn in de benen);
- visuele stoornissen;
- oogontsteking;
- ontsteking van de oogleden en zwelling van de ogen;
- duizeligheid;
- gevoel van snelle hartslag;
- hypertensie;
- opvliegers;
- hematoom;
- hoest;
- astma;
- kortademigheid;
- maagbloeding;
- dyspepsie (indigestie, opgeblazen gevoel, brandend maagzuur);
- zure refluxstoornis;
- sicca-syndroom (inclusief droge ogen en mond);
- jeuk;
- jeukende uitslag;
- blauwe plek;
- ontsteking van de huid (zoals eczeem);
- breken van de nagels van de vingers en tenen;
- toegenomen zweten;
- haaruitval;
- begin of verergering van psoriasis;
- spiertrekkingen;
- bloed in de urine;
- nierproblemen;
- pijn op de borst;
- oedeem;
- koorts;
- vermindering van bloedplaatjes in de sanggue, wat het risico op bloedingen of blauwe plekken verhoogt;
- moeite met genezen.
Soms (komen voor bij minder dan 1 op de 100 gebruikers):
- opportunistische infecties (waaronder tuberculose en andere infecties die optreden wanneer de immuunafweer is verminderd);
- neurologische infecties (inclusief virale meningitis);
- ooginfecties;
- bacteriële infecties;
- diverticulitis (ontsteking en infectie van de dikke darm);
- tumoren;
- tumoren van het lymfestelsel;
- melanoma;
- stoornissen van het immuunsysteem die de longen, huid en lymfeklieren kunnen aantasten (meestal weergegeven als sarcoïdose);
- vasculitis (ontsteking van de bloedvaten);
- tremor;
- hartinfarct;
- neuropathie;
- dubbel zicht;
- gehoorverlies, rinkelen;
- gevoel van onregelmatige hartslag zoals hartkloppingen;
- hartproblemen die kortademigheid of zwelling van de enkels kunnen veroorzaken;
- acuut myocardinfarct;
- vorming van een zak in de wand van een hoofdslagader, ontsteking en stolsel in een ader, verstopping van een bloedvat;
- longziekte die kortademigheid veroorzaakt (inclusief ontsteking);
- longembolie (afsluiting van een longslagader);
- pleurale effusie (abnormale ophoping van vocht in de pleurale ruimte);
- ontsteking van de alvleesklier die hevige pijn in de buik en rug veroorzaakt;
- moeite met slikken;
- gezichtsoedeem;
- ontsteking van de galblaas, galblaasstenen;
- dikke lever;
- Nacht zweet;
- litteken;
- abnormaal spierkatabolisme;
- systemische lupus erythematodes (inclusief ontsteking van de huid, het hart, de longen, de gewrichten en andere organen)
- onderbroken slaap;
- impotentie;
- ontstekingen.
Zelden (komen voor bij minder dan 1 op de 1.000 gebruikers):
- leukemie (kwaadaardig neoplasma dat het hematopoëtische systeem op perifeer niveau (bloed) en beenmerg aantast);
- ernstige allergische reactie met shock;
- multiple sclerose;
- neurologische aandoeningen (zoals ontsteking van de oogzenuw en syndroom van Guillain-Barré die spierzwakte, abnormale sensaties, tintelingen in de armen en het bovenlichaam kunnen veroorzaken);
- hartstilstand;
- longfibrose (littekens in de longen);
- darmperforatie;
- hepatitis;
- reactivering van hepatitis B;
- auto-immuunhepatitis (leverontsteking veroorzaakt door uw eigen immuunsysteem);
- cutane vasculitis (ontsteking van de bloedvaten in de huid);
- Stevens-Johnson-syndroom (vroege symptomen zijn onder meer malaise, koorts, hoofdpijn en huiduitslag);
- gezichtsoedeem geassocieerd met allergische reacties;
- erythema multiforme (inflammatoire huiduitslag);
- lupusachtig syndroom.
Niet bekend (frequentie kan met de beschikbare gegevens niet worden bepaald):
- hepato-milt T-cellymfoom (een zeldzame bloedkanker die vaak fataal is);
- Merkelcelcarcinoom (een type huidkanker);
- Leverfalen;
- verergering van een aandoening die dermatomyositis wordt genoemd (die zich manifesteert als huiduitslag die gepaard gaat met spierzwakte).
Sommige van de bijwerkingen die bij Humira worden waargenomen, kunnen asymptomatisch zijn en kunnen alleen in bloedonderzoek worden gevonden. Waaronder:
Zeer vaak (kan voorkomen bij meer dan 1 op de 10 mensen):
- laag aantal witte bloedcellen;
- laag aantal rode bloedcellen;
- verhoogde bloedlipiden;
- verhoogde leverenzymen.
Vaak (komen voor bij minder dan 1 op de 10 gebruikers):
- verhoogd aantal witte bloedcellen;
- verminderd aantal bloedplaatjes;
- verhoogd urinezuur in het bloed;
- verandering van natrium in het bloed;
- vermindering van calcium in het bloed;
- vermindering van fosfor in het bloed;
- verhoogde bloedsuikerspiegel;
- verhoogde bloedlactaatdehydrogenase;
- aanwezigheid van auto-antilichamen in het bloed.
Zelden (komen voor bij minder dan 1 op de 1.000 gebruikers):
- laag aantal witte bloedcellen, rode bloedcellen en bloedplaatjes.
Niet bekend (frequentie kan met de beschikbare gegevens niet worden bepaald):
- Leverfalen.
Melding van bijwerkingen
Krijgt u last van bijwerkingen, neem dan contact op met uw arts of apotheker.Dit geldt ook voor mogelijke bijwerkingen die niet in deze bijsluiter staan.
U kunt bijwerkingen ook rechtstreeks melden via het nationale meldsysteem zoals vermeld in aanhangsel V. Door bijwerkingen te melden, kunt u ons helpen meer informatie te verkrijgen over de veiligheid van dit geneesmiddel.
Vervaldatum en retentie
Buiten het zicht en bereik van kinderen houden.
Gebruik dit geneesmiddel niet meer na de uiterste houdbaarheidsdatum. Die is te vinden op het etiket/blister/doos na EXP.De uiterste houdbaarheidsdatum verwijst naar de laatste dag van die maand.
Bewaren in de koelkast (2 ° C - 8 ° C). Niet bevriezen.
Bewaar de spuit in de juiste verpakking om het geneesmiddel tegen licht te beschermen.
Alternatieve bewaarcondities:
Indien nodig (bijvoorbeeld op reis) kan een enkele gebruiksklare spuit maximaal 14 dagen bij kamertemperatuur (tot 25 ° C) worden bewaard - bescherm het geneesmiddel tegen licht. bij kamertemperatuur worden bewaard, moet de spuit binnen 14 dagen worden gebruikt of worden weggegooid, ook als deze weer in de koelkast wordt bewaard.
U moet de datum noteren waarop de spuit voor het eerst uit de koelkast is gehaald en de datum waarop de spuit moet worden weggegooid.
Geneesmiddelen niet weggooien via het afvalwater of met huishoudelijk afval Vraag uw arts of apotheker wat u met geneesmiddelen moet doen die u niet meer gebruikt, dit helpt het milieu te beschermen.
Samenstelling en farmaceutische vorm
Wat bevat Humira
Het werkzame bestanddeel is adalimumab.
De andere stoffen in dit middel zijn mannitol, citroenzuurmonohydraat, natriumcitraat, natriumdiwaterstoffosfaatdihydraat, dinatriumfosfaatdihydraat, natriumchloride, polysorbaat 80, natriumhydroxide en water voor injecties.
Dit geneesmiddel bevat minder dan 1 mmol natrium (23 mg) per dosis van 0,8 ml, daarom is het in wezen "natriumvrij" en bevat het geen conserveermiddelen.
Hoe ziet de Humira voorgevulde spuit eruit en wat is de inhoud van de verpakking
Humira voorgevulde spuit bestaat uit een oplossing van adalimumab in een glazen spuit met naaldbeveiliging Elke verpakking bevat 1 voorgevulde spuit met naaldbeveiliging voor ziekenhuisgebruik of voor toediening door derden, met 1 alcoholdoekje.
Humira 40 mg oplossing voor injectie in voorgevulde spuiten wordt geleverd als een steriele oplossing van 40 mg adalimumab opgelost in 0,8 ml oplossing.
Mogelijk worden niet alle verpakkingsgrootten in de handel gebracht.
Humira is verkrijgbaar in een injectieflacon, een voorgevulde spuit en een voorgevulde pen.
+ Bron Bijsluiter: AIFA (Italiaans Geneesmiddelenbureau). Inhoud gepubliceerd in januari 2016. De aanwezige informatie is mogelijk niet up-to-date.
Om toegang te hebben tot de meest actuele versie, is het raadzaam om de website van AIFA (Italian Medicines Agency) te bezoeken. Disclaimer en nuttige informatie.
01.0 NAAM VAN HET GENEESMIDDEL
HUMIRA 400 MG OPLOSSING VOOR INJECTIE IN VOORGEVULDE PEN
02.0 KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Elke voorgevulde pen voor eenmalig gebruik van 0,8 ml bevat 40 mg adalimumab.
Adalimumab is een recombinant humaan monoklonaal antilichaam dat tot expressie wordt gebracht in ovariumcellen van Chinese hamsters.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
03.0 FARMACEUTISCHE VORM
Heldere oplossing voor injectie in een voorgevulde pen.
04.0 KLINISCHE INFORMATIE
04.1 Therapeutische indicaties
Reumatoïde artritis
Humira, in combinatie met methotrexaat, is geïndiceerd voor:
§ Behandeling van volwassen patiënten met matige tot ernstige actieve reumatoïde artritis wanneer de respons op disease modifying anti-reumatic drugs (DMARD's), waaronder methotrexaat, onvoldoende is.
§ de behandeling van ernstige, actieve en progressieve reumatoïde artritis bij volwassenen die niet eerder met methotrexaat zijn behandeld.
Humira kan als monotherapie worden gegeven in geval van intolerantie voor methotrexaat of wanneer voortzetting van de behandeling met methotrexaat ongepast is.
Humira, in combinatie met methotrexaat, remt de progressie van radiografisch vastgestelde structuurschade en verbetert het fysieke functioneren bij deze patiëntenpopulatie.
Juveniele idiopathische artritis
Polyarticulaire juveniele idiopathische artritis
Humira in combinatie met methotrexaat is geïndiceerd voor de behandeling van actieve polyarticulaire juveniele idiopathische artritis bij patiënten vanaf 2 jaar die onvoldoende hebben gereageerd op een of meer disease-modifying anti-reumatic drugs (DMARD's).Humira kan als monotherapie worden toegediend. in geval van intolerantie voor methotrexaat of wanneer voortzetting van de behandeling met methotrexaat niet geschikt is (voor werkzaamheid bij monotherapie zie rubriek 5.1). Humira is niet onderzocht bij patiënten jonger dan 2 jaar.
Artritis geassocieerd met enthesitis
Humira is geïndiceerd voor de behandeling van actieve artritis geassocieerd met enthesitis bij patiënten van 6 jaar en ouder die onvoldoende reageerden of conventionele therapie niet verdragen (zie rubriek 5.1).
Axiale spondyloartritis
Spondylitis ankylopoetica (AS)
Humira is geïndiceerd voor de behandeling van volwassen patiënten met ernstige actieve spondylitis ankylopoetica bij wie de respons op conventionele therapie onvoldoende was.
Axiale spondyloartritis zonder radiografisch bewijs van SA
Humira is geïndiceerd voor de behandeling van volwassen patiënten met ernstige axiale spondyloartritis zonder radiografisch bewijs van AS, maar met objectieve tekenen van ontsteking gedetecteerd door verhoogd reactief proteïne C en/of MRI, die onvoldoende hebben gereageerd op, of intolerant zijn voor niet-steroïde ontstekingsremmende medicijnen.
Psoriatische arthritis
Humira is geïndiceerd voor de behandeling van actieve en progressieve artritis psoriatica bij volwassenen wanneer de respons op eerdere ziektemodificerende antireumatische geneesmiddelen (DMARD's) onvoldoende was. schade gedetecteerd door röntgenfoto's bij patiënten met symmetrische polyarticulaire subgroepen van de ziekte (zie rubriek 5.1) en verbetert de fysieke functie.
Psoriasis
Humira is geïndiceerd voor de behandeling van matige tot ernstige chronische plaque psoriasis bij volwassen patiënten die niet hebben gereageerd op, of die contra-indicaties hebben, of die intolerant zijn geweest voor andere systemische therapieën, waaronder behandeling met ciclosporine, methotrexaat of PUVA.
ziekte van Crohn
Humira is geïndiceerd voor de behandeling van matige tot ernstige actieve ziekte van Crohn bij volwassen patiënten die niet hebben gereageerd op een volledige en adequate kuur met corticosteroïden en/of een immunosuppressivum, of bij patiënten die dergelijke therapieën niet verdragen of die medische contra-indicaties hebben voor hen.
Ziekte van Crohn bij pediatrische patiënten
Humira is geïndiceerd voor de behandeling van ernstige actieve ziekte van Crohn bij pediatrische patiënten (vanaf 6 jaar) die onvoldoende reageerden op conventionele therapie ≤ waaronder primaire voedingstherapie, behandeling met corticosteroïden en een immunomodulator, of die intolerant zijn of contra-indicaties hebben voor dergelijke therapieën.
Colitis ulcerosa
Humira is geïndiceerd voor de behandeling van matige tot ernstige actieve colitis ulcerosa bij volwassen patiënten die onvoldoende reageerden op conventionele therapie, waaronder corticosteroïden en 6-mercaptopurine (6-MP) of azathioprine (AZA), of die dergelijke therapieën.
04.2 Dosering en wijze van toediening
Dosering
De behandeling met Humira moet worden gestart en gecontroleerd door gespecialiseerde artsen die ervaring hebben met de diagnose en behandeling van de aandoeningen waarvoor Humira is geïndiceerd. Patiënten die met Humira worden behandeld, moeten een speciale waarschuwingskaart krijgen.
Na de juiste instructie over de Humira-injectietechniek, kunnen patiënten zichzelf injecteren als hun arts dit nodig acht, en indien nodig met periodieke medische controles.
Tijdens de behandeling met Humira moeten andere gelijktijdige therapieën (bijvoorbeeld corticosteroïden en/of immunomodulerende middelen) worden geoptimaliseerd.
Reumatoïde artritis
De dosis Humira die is geïndiceerd voor volwassen patiënten met reumatoïde artritis is 40 mg adalimumab als een enkele subcutane injectie om de twee weken.Methotrexaat moet worden voortgezet tijdens de behandeling met Humira.
Glucocorticoïden, salicylaten, niet-steroïdale anti-inflammatoire geneesmiddelen of analgetica kunnen tijdens de behandeling met Humira worden voortgezet. Zie rubrieken 4.4 en 5.1 voor de combinatie met andere DMARD's dan methotrexaat.
Sommige patiënten die een verminderde respons vertonen op monotherapie, kunnen baat hebben bij een verhoging van hun dosis tot wekelijks 40 mg adalimumab.
Stopzetting van de dosis
Het kan nodig zijn om de toediening te onderbreken, bijvoorbeeld vóór een operatie of in geval van een ernstige infectie.
Beschikbare gegevens geven aan dat herintroductie van Humira na stopzetting van 70 dagen of langer resulteert in een klinische respons van hetzelfde belang en met een vergelijkbaar veiligheidsprofiel als vóór stopzetting van de dosering.
Spondylitis ankylopoetica, axiale spondyloartritis zonder röntgenologisch bewijs van AS en artritis psoriatica
De aanbevolen dosis Humira voor patiënten met spondylitis ankylopoetica, axiale spondylartritis zonder röntgenologisch bewijs van AS en voor patiënten met artritis psoriatica is 40 mg adalimumab om de twee weken toegediend als een enkelvoudige dosis subcutaan.
Voor alle bovengenoemde indicaties suggereren de beschikbare gegevens dat klinische respons gewoonlijk wordt bereikt binnen 12 weken na aanvang van de behandeling.In gevallen waarin binnen deze periode geen respons optreedt, dient voortzetting van de behandeling zorgvuldig te worden overwogen.
Psoriasis
De aanbevolen dosis Humira voor volwassen patiënten is een startdosis van 80 mg, subcutaan toegediend, gevolgd door een dosis van 40 mg, subcutaan, eenmaal per twee weken, beginnend in de week volgend op het innemen van de startdosis.
Er moet zorgvuldig worden overwogen of de behandeling langer dan 16 weken moet worden voortgezet als patiënten binnen deze periode geen bevredigende respons hebben ontwikkeld.
ziekte van Crohn
De aangegeven dosis Humira voor inductietherapie is 80 mg in week 0 voor volwassen patiënten met matige tot ernstige ziekte van Crohn, gevolgd door 40 mg in week 2. Als een snellere respons op de therapie nodig is, kan een dosis van 160 mg in week 0 worden gegeven (deze dosis kan worden gegeven als vier injecties in de loop van een dag of twee injecties per dag gedurende twee opeenvolgende dagen), gevolgd door 80 mg in week 2, rekening houdend met het feit dat het risico op bijwerkingen groter is tijdens inductie.
Na inductiebehandeling is de aangegeven dosis 40 mg eenmaal per twee weken, subcutaan toegediend. Als een patiënt de behandeling met Humira heeft stopgezet en de ziektesymptomen terugkeren, kan de behandeling met Humira ook opnieuw worden toegediend. Er zijn weinig gegevens over het opnieuw toedienen van Humira als er een periode van 8 weken is verstreken sinds de vorige dosis werd gegeven.
Tijdens onderhoudstherapie kan de dosering van corticosteroïden geleidelijk worden verlaagd volgens richtlijnen die zijn ontwikkeld voor klinische behandeling van de ziekte.
Sommige patiënten bij wie de respons op de therapie verminderd is, kunnen baat hebben bij een verhoging van de doseringsfrequentie tot wekelijks 40 mg Humira.
Patiënten die tegen week 4 geen adequate respons op de therapie hebben laten zien, kunnen baat hebben bij voortzetting van de onderhoudstherapie tot en met week 12. Bij patiënten bij wie de respons op de therapie binnen deze tijd onvoldoende is, moet zorgvuldig worden beoordeeld of voortzetting van de therapie noodzakelijk is.
Colitis ulcerosa
Het aanbevolen inductiedoseringsschema van Humira voor volwassen patiënten met matige tot ernstige colitis ulcerosa is 160 mg in week 0 (de dosis kan worden toegediend als 4 injecties op één dag of als twee injecties per dag, gedurende twee dagen). mg per week 2. Na inductiebehandeling is de aanbevolen dosis 40 mg eenmaal per twee weken subcutaan.
Tijdens de onderhoudsbehandeling kunnen corticosteroïden geleidelijk worden afgebouwd volgens klinische praktijkrichtlijnen.
Patiënten met een gedocumenteerde verminderde respons kunnen baat hebben bij een verhoging van de doseringsfrequentie tot wekelijks 40 mg Humira.
Beschikbare gegevens suggereren dat klinische respons gewoonlijk binnen 2-8 weken na behandeling wordt bereikt.
De behandeling met Humira mag niet worden voortgezet bij patiënten die gedurende deze tijd niet hebben gereageerd.
Ouderen
Er zijn geen dosisaanpassingen nodig.
Lever- en/of nierinsufficiëntie
Humira is niet onderzocht bij deze patiëntenpopulaties. Er kunnen geen doseringsadviezen worden gegeven.
Pediatrische populatie
Juveniele idiopathische artritis
Polyarticulaire juveniele idiopathische artritis van 2 tot 12 jaar
De aanbevolen dosis Humira voor patiënten met polyarticulaire juveniele idiopathische artritis van 2 tot 12 jaar is 24 mg/m2 lichaamsoppervlak tot een maximale enkelvoudige dosis van 20 mg adalimumab (voor patiënten van 2 jaar - lengte en gewicht van de patiënt (tabel 1) Er is een pediatrische injectieflacon van 40 mg beschikbaar voor patiënten die een dosering onder de maximale dosis van 40 mg moeten nemen.
Tabel 1. Humira-dosis in milliliter (ml) naar lengte en gewicht bij patiënten met polyarticulaire juveniele idiopathische artritis en enthesitis-geassocieerde artritis
* De maximale enkelvoudige dosis is 40 mg (0,8 ml)
Polyarticulaire juveniele idiopathische artritis vanaf 13 jaar
Voor patiënten van 13 jaar en ouder wordt om de twee weken een dosis van 40 mg toegediend, ongeacht het lichaamsoppervlak.
Beschikbare gegevens suggereren dat klinische respons gewoonlijk binnen 12 weken na behandeling wordt bereikt. Bij patiënten bij wie de respons op de therapie binnen deze periode onvoldoende is, moet de noodzaak van voortzetting van de therapie zorgvuldig worden overwogen.
Er is geen relevante toepassing van Humira bij patiënten jonger dan 2 jaar voor deze indicatie.
Artritis geassocieerd met enthesitis
De aanbevolen dosis Humira bij patiënten met enthesitis-geassocieerde artritis van 6 jaar en ouder is 24 mg/m2 lichaamsoppervlak, tot een maximale enkelvoudige dosis van 40 mg adalimumab eenmaal per twee weken toegediend via subcutane injectie. Het injectievolume wordt gekozen op basis van de lengte en het gewicht van de patiënt (tabel 1).
Humira is niet onderzocht bij patiënten jonger dan 6 jaar met enthesitis-geassocieerde artritis.
Pediatrische psoriasis
De veiligheid en werkzaamheid van Humira bij kinderen van 4-17 jaar zijn niet vastgesteld Er zijn geen gegevens beschikbaar Er is geen relevante toepassing van Humira bij kinderen jonger dan 4 jaar voor deze indicatie.
Ziekte van Crohn bij pediatrische patiënten
Ziekte van Crohn bij pediatrische patiënten
De aanbevolen inductiedosis Humira bij pediatrische patiënten met ernstige ziekte van Crohn is 40 mg in week 0, gevolgd door 20 mg in week 2. Als een snellere respons op de therapie nodig is, kan een regime van 80 mg in week 0 worden gebruikt. ( dosis kan worden gegeven als twee injecties op één dag) en 40 mg in week 2, met dien verstande dat het risico op bijwerkingen hoger kan zijn bij gebruik van de hogere inductiedosis.
Na inductiebehandeling is de aanbevolen dosis 20 mg eenmaal per twee weken via een subcutane injectie. Sommige personen met onvoldoende respons kunnen baat hebben bij een verhoging van de doseringsfrequentie tot wekelijks 20 mg Humira.
Ziekte van Crohn bij pediatrische patiënten ≥ 40 kg:
De aanbevolen inductiedosis Humira bij pediatrische patiënten met ernstige ziekte van Crohn is 80 mg in week 0, gevolgd door 40 mg in week 2. Als een snellere respons op de therapie nodig is, kan een regime van 160 mg in week 0 worden gebruikt. ( de dosis kan worden gegeven als vier injecties op één dag of als twee injecties per dag gedurende twee opeenvolgende dagen) en 80 mg in week 2, met dien verstande dat het risico op bijwerkingen hoger kan zijn bij gebruik van de hogere dosis inductie.
Na de inductiebehandeling is de aanbevolen dosis 40 mg eenmaal per twee weken via een subcutane injectie. Sommige personen met onvoldoende respons kunnen baat hebben bij een verhoging van de dosisfrequentie tot 40 mg Humira per week.
Voortzetting van de behandeling moet zorgvuldig worden overwogen bij een proefpersoon die in week 12 nog niet reageert.
Er is geen relevante toepassing van Humira bij kinderen van 6 jaar en jonger voor deze indicatie.
Colitis ulcerosa bij kinderen
De veiligheid en werkzaamheid van Humira bij kinderen van 4-17 jaar zijn nog niet vastgesteld Er zijn geen gegevens beschikbaar Er is geen relevante toepassing van Humira bij kinderen jonger dan 4 jaar voor deze indicatie.
Artritis psoriatica en axiale spondyloartritis inclusief spondylitis ankylopoetica
Er is geen relevante toepassing van Humira bij pediatrische patiënten voor de indicaties spondylitis ankylopoetica en artritis psoriatica.
Wijze van toediening
Humira wordt toegediend via een injectie onder de huid. De volledige gebruiksaanwijzing vindt u in de bijsluiter.
Een pediatrische injectieflacon van 40 mg is beschikbaar voor patiënten die toediening van minder dan de volledige dosis van 40 mg nodig hebben.
04.3 Contra-indicaties
Overgevoeligheid voor de werkzame stof of voor één van de in rubriek 6.1 vermelde hulpstoffen.
Actieve tuberculose of andere ernstige infecties zoals sepsis en opportunistische infecties (zie rubriek 4.4).
Matig tot ernstig hartfalen (NYHA klasse III/IV) (zie rubriek 4.4).
04.4 Bijzondere waarschuwingen en passende voorzorgen bij gebruik
Om de traceerbaarheid van biologische geneesmiddelen te verbeteren, moeten het handelsmerk en het batchnummer van het toegediende product duidelijk worden vastgelegd (of gemarkeerd).
infecties
Patiënten die worden behandeld met TNF-antagonisten zijn vatbaarder voor ernstige infecties. Een verminderde longfunctie kan het risico op het ontwikkelen van infecties vergroten.
Patiënten moeten daarom vóór, tijdens en na de behandeling met Humira zorgvuldig worden gescreend op infecties, waaronder tuberculose. Aangezien de eliminatie van adalimumab tot vier maanden kan duren, moet de controle gedurende deze periode worden voortgezet.
Behandeling met Humira mag niet worden gestart bij patiënten met actieve infecties, waaronder chronische of gelokaliseerde infecties, totdat deze onder controle zijn. Bij patiënten die zijn blootgesteld aan tuberculose en bij patiënten die zijn gereisd naar gebieden met een hoog risico op tuberculose of endemische mycose, zoals histoplasmose, coccidioidomycose of blastomycose, moeten de risico's en voordelen van behandeling met Humira worden afgewogen voordat de behandeling wordt gestart (zie voor meer informatie over Opportunistische infecties).
Patiënten die tijdens de behandeling met Humira een nieuwe infectie ontwikkelen, moeten nauwlettend worden gevolgd en een volledige diagnostische evaluatie ondergaan. Als zich een nieuwe ernstige infectie of sepsis ontwikkelt, moet de toediening van Humira worden gestaakt en moet een geschikte antimicrobiële of antischimmeltherapie worden ingesteld totdat de infectie onder controle is. patiënten vatbaar maken voor infecties, waaronder gelijktijdig gebruik van immunosuppressiva.
Ernstige infecties:
Er zijn meldingen geweest van ernstige infecties, waaronder sepsis, veroorzaakt door bacteriën, mycobacteriën, invasieve schimmels, parasieten, virussen of andere opportunistische infecties zoals listeriose, legionellose en pneumocystose bij patiënten die met Humira werden behandeld.
Andere ernstige infecties die in klinische onderzoeken zijn waargenomen, zijn onder meer longontsteking, pyelonefritis, septische artritis en bloedvergiftiging. Gevallen van ziekenhuisopname of dodelijke voorvallen in verband met infecties zijn gemeld.
Tuberculose:
Tuberculose, waaronder reactivering en het ontstaan van tuberculose, is gemeld bij patiënten die Humira gebruikten. Er zijn gevallen van pulmonale en extrapulmonale (d.w.z. gedissemineerde) tuberculose gemeld.
Voordat de behandeling met Humira wordt gestart, moeten alle patiënten worden onderzocht op de aanwezigheid van actieve of inactieve ("latente") tuberculose. Deze evaluatie moet een "gedetailleerde medische geschiedenis omvatten van patiënten met een voorgeschiedenis van tuberculose of enig contact met mensen met actieve tuberculose en met eerdere en / of gelijktijdige immunosuppressieve therapieën. Passende screeningtests (dwz huidtest met tuberculine en thoraxfoto's) ) bij alle patiënten (lokale richtlijnen kunnen worden gevolgd) Het wordt aanbevolen om deze tests uit te voeren en de resultaten te noteren op de patiëntenwaarschuwingskaart. Artsen moeten alert zijn op het risico van vals-negatieve resultaten van tuberculinehuidtesten, vooral bij ernstig zieke of immuungecompromitteerde patiënten.
Als actieve tuberculose wordt gediagnosticeerd, mag de behandeling met Humira niet worden gestart (zie rubriek 4.3).
In alle hieronder beschreven situaties moet een zorgvuldige evaluatie van de risico-batenverhouding van Humira-therapie worden uitgevoerd.
Bij een vermoeden van latente tuberculose is het raadzaam een arts te raadplegen die gespecialiseerd is in de behandeling van tuberculose.
Als latente tuberculose wordt gediagnosticeerd, moet een antituberculoseprofylaxebehandeling worden ingesteld volgens de lokale aanbevelingen voordat de behandeling met Humira wordt gestart.
De instelling van een antituberculoseprofylaxebehandeling dient ook te worden overwogen voordat de behandeling met Humira wordt gestart bij patiënten met verschillende of significante risicofactoren voor tuberculose ondanks een negatieve test op tuberculose en bij die patiënten bij wie in hun medische voorgeschiedenis een persoonlijke voorgeschiedenis van latente of actieve tuberculose bij waarvan niet kan worden vastgesteld of de behandeling die zij hebben ondergaan adequaat was.
Ondanks profylactische behandeling van tuberculose is reactivering van tuberculose opgetreden bij patiënten die met Humira werden behandeld. Sommige patiënten die met succes werden behandeld voor actieve tuberculose, kregen opnieuw tuberculose tijdens de behandeling met Humira.
Patiënten moeten het advies krijgen om medische hulp in te roepen als zich tijdens of na de behandeling met Humira tekenen/symptomen voordoen die wijzen op een mogelijke tuberculeuze infectie (bijv. aanhoudende hoest, verspilling, gewichtsverlies, matige koorts, lusteloosheid).
Andere opportunistische infecties:
Gevallen van opportunistische infecties, waaronder invasieve schimmelinfecties, zijn waargenomen bij patiënten die Humira gebruiken. Deze infecties zijn niet correct gediagnosticeerd bij patiënten die TNF-antagonisten gebruiken en dit heeft geleid tot een vertraging in de juiste behandeling, soms met fatale afloop.
Bij patiënten die tekenen en symptomen ontwikkelen zoals koorts, malaise, gewichtsverlies, zweten, hoesten, dyspneu en/of longinfiltraat of een andere ernstige systemische ziekte met of zonder gelijktijdige shock, moet een invasieve schimmelinfectie worden vermoed en onmiddellijk worden stopgezet. Humira Diagnose en toediening van empirische antischimmeltherapie bij deze patiënten moet worden gemaakt in overleg met een arts die gespecialiseerd is in de behandeling van patiënten met invasieve schimmelinfecties.
Reactivering van hepatitis B
Reactivering van hepatitis B (bijv. positief oppervlakte-antigeen) is opgetreden bij chronische dragers van het hepatitis B-virus die werden behandeld met TNF-antagonisten, waaronder Humira. Sommige gevallen hebben een fatale afloop gehad. Voordat de behandeling met Humira wordt gestart, moeten patiënten worden getest op infectie met het hepatitis B-virus. Bij patiënten die positief testen op het hepatitis B-virus, wordt aangeraden een arts te raadplegen die ervaring heeft met de behandeling van hepatitis B.
Dragers van het hepatitis B-virus die behandeling met Humira nodig hebben, moeten nauwlettend worden gecontroleerd op tekenen en symptomen van een actieve infectie met het hepatitis B-virus, niet alleen gedurende de hele behandeling, maar ook gedurende de maanden na het staken van de behandeling. Er zijn geen adequate gegevens beschikbaar over de behandeling van patiënten met hepatitis B-virus die antivirale therapie ondergaan om reactivering van het hepatitis B-virus gelijktijdig met therapie met TNF-antagonisten te voorkomen. Bij patiënten die reactivering van het hepatitis B-virus ontwikkelen, moet de toediening van Humira worden gestaakt en moet een effectieve antivirale therapie worden ingesteld, vergezeld van adequate ondersteunende behandeling.
Neurologische gebeurtenissen
TNF-antagonisten, waaronder Humira, zijn in zeldzame gevallen in verband gebracht met het ontstaan of verergeren van klinische symptomen en/of radiografisch bewijs van demyeliniserende ziekten van het centrale zenuwstelsel, waaronder multiple sclerose, optische neuritis en perifere demyeliniserende ziekten, waaronder het syndroom van Guillain-Barrè. Voorzichtigheid is geboden bij het gebruik van Humira bij patiënten met eerdere of recente aanvang van demyeliniserende aandoeningen van het centrale of perifere zenuwstelsel.
Allergische reacties
In klinische onderzoeken waren ernstige allergische reacties geassocieerd met Humira zeldzaam. Niet-ernstige allergische reacties geassocieerd met Humira tijdens klinische onderzoeken kwamen soms voor. Er zijn meldingen geweest van ernstige allergische reacties, waaronder anafylaxie na toediening van Humira.Als anafylactische reacties of andere ernstige allergische verschijnselen optreden, moet de toediening van Humira onmiddellijk worden gestaakt en moet een passende therapie worden gestart.
Immunosuppressie
In een onderzoek onder 64 patiënten met reumatoïde artritis die werden behandeld met Humira, was er geen bewijs van remming van vertraagde overgevoeligheid, verlaging van immunoglobulinespiegels of veranderingen in het aantal T-, B-, NK-, monocyt-/cellymfocyten, macrofagen en neutrofielen.
Neoplasmata en lymfoproliferatieve ziekten
In de gecontroleerde secties van de klinische onderzoeken met TNF-antagonisten werden meer gevallen van maligniteiten, waaronder lymfoom, waargenomen bij patiënten die TNF-blokkers kregen dan in de controlegroep. Gevallen waren echter zeldzaam. In postmarketingonderzoeken zijn gevallen van leukemie gemeld bij patiënten die werden behandeld met een TNF-antagonist. Er is een groter verhoogd risico op het ontwikkelen van lymfomen en leukemie bij patiënten met ernstig actieve en langdurige reumatoïde artritis, een ontstekingsziekte die de risicobeoordeling bemoeilijkt Met de huidige kennis kan de ontwikkeling van lymfomen niet worden uitgesloten leukemie en andere maligniteiten bij patiënten behandeld met anti-TNF-medicijnen.
Gevallen van kanker, waarvan sommige fataal, zijn gemeld bij kinderen, adolescenten en jonge volwassenen (tot 22 jaar) die werden behandeld met TNF-antagonisten (start van de behandeling ≤ 18 jaar), waaronder adalimumab in postmarketingonderzoeken. Ongeveer de helft van de gevallen waren lymfomen. De andere gevallen vertegenwoordigden een veelvoud van verschillende kankers en omvatten zeldzame kankers die gewoonlijk worden geassocieerd met immunosuppressie. Een risico op het ontstaan van tumoren bij kinderen en adolescenten die met TNF-antagonisten worden behandeld, kan niet worden uitgesloten.
Zeldzame postmarketinggevallen van hepatosplenisch T-cellymfoom zijn waargenomen bij patiënten die werden behandeld met adalimumab. Dit zeldzame type T-cellymfoom heeft een zeer agressief klinisch beloop en is vaak fataal. Sommige van deze gevallen van hepatosplenisch T-cellymfoom traden op bij jongvolwassen patiënten die werden behandeld met Humira en die gelijktijdig werden behandeld met azathioprine of 6-mercaptopurine, geneesmiddelen die worden gebruikt voor de behandeling van inflammatoire darmaandoeningen. Het potentiële risico van de combinatie van azathioprine of 6-mercaptopurine en Humira moet zorgvuldig worden overwogen. Een risico op het ontwikkelen van hepatosplenisch T-cellymfoom kan niet worden uitgesloten bij patiënten die met Humira worden behandeld (zie rubriek 4.8).
Er zijn geen klinische onderzoeken uitgevoerd bij patiënten met een voorgeschiedenis van kanker of bij patiënten bij wie de behandeling met Humira werd voortgezet na de ontwikkeling van kanker. Daarom moet behandeling met Humira bij deze patiëntenpopulatie met extra voorzichtigheid worden overwogen (zie rubriek 4.8).
Voor en tijdens de behandeling met Humira moeten alle patiënten, met name degenen met een voorgeschiedenis van uitgebreide immunosuppressieve therapie of degenen met psoriasis die een voorgeschiedenis hebben van behandeling met PUVA, worden onderzocht op de aanwezigheid van een mogelijke niet-melanotische huidkanker. Melanoom en Merkelcelcarcinoom zijn ook gemeld bij patiënten die werden behandeld met TNF-antagonisten, waaronder adalimumab (zie rubriek 4.8).
In een verkennend klinisch onderzoek naar het gebruik van een andere TNF-antagonist, infliximab, bij patiënten met matige tot ernstige chronische obstructieve longziekte (COPD), werden meer maligniteiten gemeld bij patiënten die met infliximab werden behandeld dan bij controlepatiënten, vooral in de longen of het hoofd. en nek Alle patiënten hadden een voorgeschiedenis van zware rokers.
Daarom is voorzichtigheid geboden bij het gebruik van een TNF-antagonist bij COPD-patiënten, evenals bij patiënten met een verhoogd risico op maligniteit als gevolg van overmatig roken.
Op basis van de huidige gegevens is het niet bekend of behandeling met adalimumab het risico op het ontwikkelen van dysplasie of darmkanker beïnvloedt. Alle patiënten met colitis ulcerosa die een verhoogd risico hebben op colondysplasie of carcinoom (bijvoorbeeld patiënten met langdurige colitis ulcerosa of primaire scleroserende cholangitis), of die een voorgeschiedenis van dysplasie of kanker van het colon colon hebben gehad, moeten worden gescreend regelmatig voor dysplasie in de loop van de ziekte. Deze evaluatie moet coloscopieën en biopsieën omvatten op basis van lokale aanbevelingen.
Reacties die het hematopoëtische systeem beïnvloeden
Zeldzame gevallen van pancytopenie, waaronder het optreden van aplastische anemie, zijn gemeld na het gebruik van TNF-antagonisten Bijwerkingen van het hematopoëtische systeem, waaronder significante cytopenieën sinds medisch oogpunt (bijvoorbeeld trombocytopenie, leukopenie) Tijdens behandeling met Humira moeten alle patiënten worden gewezen op de noodzaak om onmiddellijk een arts te raadplegen om adequate hulp te krijgen als zich tekenen en symptomen ontwikkelen die op dyscrasie wijzen (bijv. aanhoudende koorts, blauwe plekken, bloeding, bleekheid) Bij patiënten met bevestigde significante veranderingen in de hematopoëtisch systeem, moet de noodzaak worden overwogen om de behandeling met Humira te staken.
Vaccinaties
Vergelijkbare antilichaamreacties op het standaard 23-valent pneumokokkenvaccin en het trivalente influenzavirusvaccin werden waargenomen in een onderzoek onder 226 volwassen proefpersonen met reumatoïde artritis die werden behandeld met adalimumab of placebo. Geen gegevens beschikbaar over secundaire overdracht van infectie door levende vaccins in patiënten die Humira gebruiken.
Bij pediatrische patiënten wordt aanbevolen om het geplande vaccinatieschema, indien mogelijk, te implementeren in overeenstemming met de huidige vaccinatierichtlijnen voordat een op Humira gebaseerde therapie wordt gestart.
Met Humira behandelde patiënten kunnen gelijktijdige vaccinaties krijgen, met uitzondering van levende vaccins. Toediening van levende vaccins aan zuigelingen die in utero aan adalimumab zijn blootgesteld, wordt niet aanbevolen gedurende 5 maanden na de laatste toediening van adalimumab door de moeder tijdens de zwangerschap.
Congestief hartfalen
Verergering van congestief hartfalen en de daarmee samenhangende verhoogde mortaliteit werden waargenomen in een klinische studie met een ander anti-TNF-geneesmiddel. Verergering van congestief hartfalen is ook waargenomen bij patiënten die met Humira werden behandeld. Humira moet met voorzichtigheid worden gebruikt bij patiënten met licht hartfalen (NYHA klasse I/II). Humira is gecontra-indiceerd bij matig of ernstig hartfalen (zie rubriek 4.3) Behandeling met Humira dient te worden gestaakt bij patiënten met verergering of nieuwe symptomen van congestief hartfalen.
Auto-immuunprocessen
Behandeling met Humira kan de vorming van auto-immuunantilichamen induceren. De impact van langdurige behandeling met Humira op de ontwikkeling van auto-immuunziekten is niet bekend Als een patiënt symptomen ontwikkelt die duiden op lupusachtig syndroom na behandeling met Humira en positief is voor antilichamen tegen dubbelstrengs DNA, mag de behandeling met Humira niet worden voortgezet. moet worden gegeven (zie rubriek 4.8).
Gelijktijdige toediening van biologische DMARDS of TNF-antagonisten
Ernstige infecties zonder klinisch voordeel in vergelijking met alleen etanercept zijn waargenomen in klinische onderzoeken naar combinatietherapie met anakinra en een ander anti-TNF-geneesmiddel, etanercept.
Gezien het soort bijwerkingen dat is waargenomen bij de combinatie van anakinra en etanercept, kunnen vergelijkbare bijwerkingen optreden na de combinatie van anakinra en een ander anti-TNF-geneesmiddel. Daarom wordt de combinatie van adalimumab met anakinra niet aanbevolen (zie rubriek 4.5).
Gelijktijdige toediening van adalimumab met andere biologische DMARDS (bijv. anakinra en abatacept) of andere TNF-antagonisten wordt niet aanbevolen vanwege een mogelijk verhoogd risico op infecties, waaronder ernstige infecties en andere mogelijke geneesmiddelinteracties (zie rubriek 4.5).
Chirurgische ingrepen
Er is "beperkte" ervaring bij patiënten die met Humira worden behandeld met betrekking tot de veiligheid van chirurgische ingrepen. Bij het plannen van een operatie moet rekening worden gehouden met de lange halfwaardetijd van adalimumab. Een patiënt die een operatie ondergaat terwijl hij met Humira wordt behandeld, moet nauwlettend worden gevolgd voor de ontwikkeling van infecties, in welk geval actie moet worden ondernomen. Er is "beperkte" ervaring met betrekking tot de veiligheid bij patiënten die een gewrichtsvervangende operatie ondergaan terwijl ze Humira gebruiken.
Dunne darmobstructie
Als u niet reageert op de behandeling van de ziekte van Crohn, kan dit duiden op de aanwezigheid van rigide fibrotische stenose waarvoor een operatie nodig kan zijn. Beschikbare gegevens suggereren dat Humira niet verergert of vernauwingen veroorzaakt.
Ouderen
De frequentie van ernstige infecties bij met Humira behandelde patiënten ouder dan 65 jaar (3,5%) was hoger dan bij patiënten jonger dan 65 jaar (1,5%). Sommige hiervan hebben een fatale afloop gehad. Bij de behandeling van oudere patiënten moet bijzondere aandacht worden besteed aan het risico op infectie.
Pediatrische populatie
Zie Vaccinaties hierboven.
04.5 Interacties met andere geneesmiddelen en andere vormen van interactie
Humira-therapie is onderzocht als monotherapie en in combinatie met methotrexaat bij patiënten met reumatoïde artritis, polyarticulaire juveniele idiopathische artritis en artritis psoriatica. De vorming van antilichamen was lager wanneer Humira in combinatie met methotrexaat werd gegeven dan bij monotherapie.Toediening van Humira zonder methotrexaat resulteerde in verhoogde antilichaamvorming, verhoogde klaring en verminderde werkzaamheid van adalimumab (zie rubriek 5.1).
De combinatie van Humira en anakinra wordt niet aanbevolen (zie rubriek 4.4 "Gelijktijdige toediening van biologische DMARD's of TNF-antagonisten").
De combinatie van Humira en abatacept wordt niet aanbevolen (zie rubriek 4.4 "Gelijktijdige toediening van biologische DMARD's of TNF-antagonisten").
04.6 Zwangerschap en borstvoeding
Zwangerschap
Voor Humira zijn beperkte klinische gegevens over blootgestelde zwangerschappen beschikbaar.
In een ontwikkelingstoxicologisch onderzoek bij apen werd geen maternale toxiciteit, embryotoxiciteit of teratogeniteit gevonden. Er zijn geen preklinische gegevens beschikbaar over postnatale toxiciteit van adalimumab (zie rubriek 5.3).
Vanwege TNFα-remming kan toediening van adalimumab tijdens de zwangerschap de normale immuunrespons van de pasgeborene verstoren.Adalimumab wordt daarom niet aanbevolen tijdens de zwangerschap.
Adalimumab kan de placenta passeren en het serum bereiken van baby's van moeders die tijdens de zwangerschap met adalimumab zijn behandeld. Daardoor lopen deze kinderen een groter risico op besmetting. Toediening van levende vaccins aan zuigelingen die in utero aan adalimumab zijn blootgesteld, wordt niet aanbevolen gedurende 5 maanden na de laatste toediening van adalimumab door de moeder tijdens de zwangerschap.
Voedertijd
Het is niet bekend of adalimumab na inname in de moedermelk wordt uitgescheiden of systemisch wordt geabsorbeerd.
Omdat humane immunoglobulinen echter in de melk worden uitgescheiden, dienen vrouwen gedurende ten minste vijf maanden na hun laatste behandeling met Humira geen borstvoeding te geven.
Vruchtbaarheid
Er zijn geen preklinische gegevens beschikbaar over de effecten van adalimumab op de vruchtbaarheid.
Vrouwen in de vruchtbare leeftijd. Anticonceptie bij mannen en vrouwen
Vrouwen die zwanger kunnen worden, moeten adequate anticonceptie gebruiken om zwangerschap te voorkomen en het gebruik ervan voortzetten tot ten minste vijf maanden na de laatste Humira-behandeling.
04.7 Beïnvloeding van de rijvaardigheid en het vermogen om machines te bedienen
Humira heeft geringe effecten op de rijvaardigheid of op het vermogen om machines te bedienen. Duizeligheid en visusstoornissen kunnen optreden na toediening van Humira (zie rubriek 4.8).
04.8 Bijwerkingen
Humira is gedurende maximaal 60 maanden of langer onderzocht bij 8.198 patiënten in centrale gecontroleerde en open-label klinische onderzoeken. Deze onderzoeken werden uitgevoerd bij patiënten met vroeg optredende en langdurige reumatoïde artritis, juveniele idiopathische artritis (polyarticulaire juveniele idiopathische artritis en enthesitis-geassocieerde artritis), evenals bij patiënten met axiale spondyloartritis (spondylitis ankylopoetica en axiale spondyloartritis zonder bewijs van SA), artritis psoriatica, de ziekte van Crohn, colitis ulcerosa en psoriasis. De belangrijkste gecontroleerde onderzoeken werden uitgevoerd bij 5.343 patiënten die Humira kregen en bij 3.148 patiënten die placebo of een actieve comparator kregen tijdens de controleperiode.
Het percentage patiënten dat stopte met de behandeling vanwege bijwerkingen tijdens de dubbelblinde, gecontroleerde fase van de hoofdonderzoeken was 6,1% voor patiënten die Humira gebruikten en 5,7% voor met controle behandelde patiënten.
Samenvatting van het veiligheidsprofiel
De meest gemelde bijwerkingen zijn infecties (zoals nasofaryngitis, bovenste luchtweginfectie en sinusitis), reacties op de toedieningsplaats (erytheem, pruritus, bloeding, pijn of zwelling), hoofdpijn en musculoskeletale pijn.
Er zijn ernstige bijwerkingen gemeld voor Humira. TNF-blokkerende geneesmiddelen, zoals Humira, beïnvloeden het immuunsysteem en het gebruik ervan kan de afweer van het lichaam tegen infecties en kanker beïnvloeden.
Gevallen van fatale infecties (waaronder gevallen van sepsis, ooginfecties en tuberculose), reactivering van HBV-infectie en verschillende soorten maligniteiten (waaronder gevallen van leukemie, lymfoom en hepatolymfoom) zijn ook gemeld na toediening van Humira. cellen).
Ernstige hematologische, neurologische en auto-immuunreacties zijn ook gemeld. De laatste omvatten zeldzame gevallen van pancytopenie, aplastische anemie, centrale en perifere demyelinisatie en gevallen van lupus, lupus-gerelateerde aandoeningen en Stevens-Johnson-syndroom.
Pediatrische populatie
Bijwerkingen bij pediatrische patiënten
Over het algemeen waren de bijwerkingen bij pediatrische patiënten vergelijkbaar met die bij volwassen patiënten wat betreft frequentie en type.
Tabel van de lijst met bijwerkingen
De volgende lijst van bijwerkingen is gebaseerd op ervaring uit klinische onderzoeken en postmarketingervaring en is ingedeeld naar betrokken systeem/orgaan en frequentie (zeer vaak ≥1/10; vaak ≥1/100 tot
tafel 2
Bijwerkingen
* er is meer informatie in rubrieken 4.3, 4.4 en 4.8
** inclusief open label extensieonderzoeken
1) inclusief gegevens uit spontane meldingen
Beschrijving van geselecteerde bijwerkingen
Reacties op de injectieplaats
In centrale gecontroleerde klinische onderzoeken bij volwassenen en kinderen ondervond 13,6% van de met Humira behandelde patiënten reacties op de injectieplaats (erytheem en/of pruritus, bloeding, pijn of oedeem), tegenover 7,6% van de patiënten die werden behandeld met placebo of actieve controle. was over het algemeen niet nodig om de medicatie te staken.
infecties
In de centrale gecontroleerde klinische onderzoeken bij volwassenen en kinderen was het infectiepercentage 1,52 per patiënt/jaar in de Humira-groep en 1,45 per patiënt/jaar in de placebo- en actieve controlegroepen.Infecties werden voornamelijk vertegenwoordigd door nasofaryngitis, bovenste luchtweginfecties en urineweginfectie De meeste patiënten bleven Humira gebruiken nadat de infectie was verdwenen.
De incidentie van ernstige infecties was 0,04 per patiënt/jaar in de Humira-groep en 0,03 per patiënt/jaar in de placebo- en actief gecontroleerde groepen.
In gecontroleerde en open-label onderzoeken met Humira bij volwassenen en kinderen zijn ernstige infecties (waaronder dodelijke infecties, die slechts zelden optraden), waaronder gevallen van tuberculose (inclusief miliaire en extrapulmonale locaties) en invasieve opportunistische infecties ( bijvoorbeeld van gedissemineerde of extrapulmonale histoplasmose, blastomycose, coccidioidomycose, pneumocystose, candidiasis, aspergillose en listeriose). De meeste gevallen van tuberculose traden op binnen de eerste acht maanden na aanvang van de therapie en kunnen worden geïnterpreteerd als een heropflakkering van latente ziekte.
Neoplasmata en lymfoproliferatieve ziekten
In onderzoeken die werden uitgevoerd door Humira toe te dienen aan patiënten met juveniele idiopathische artritis (polyarticulaire juveniele idiopathische artritis en enthesitis-geassocieerde artritis), werden geen maligniteiten waargenomen bij 249 pediatrische patiënten met een blootstelling van 655,6 patiëntjaren. patiënten met een blootstelling van 258,9 patiëntjaren tijdens een onderzoek waarbij Humira werd toegediend aan pediatrische patiënten met de ziekte van Crohn.
In gecontroleerde secties van hoofdonderzoeken bij volwassenen met Humira die ten minste 12 weken duurden bij patiënten met matige tot ernstige actieve reumatoïde artritis, spondylitis ankylopoetica, axiale spondyloartitis zonder radiografisch bewijs van AS, artritis psoriatica, psoriasis, ziekte van Crohn en colitis ulcerziekte, neoplasmata, zoals evenals lymfoom en niet-melanotische huidkanker, werden waargenomen met een percentage (95% betrouwbaarheidsinterval) van 6,0 (3,7; 9,8) per 1.000 patiëntjaren bij 4.622 patiënten die werden behandeld met Humira versus een percentage van 5,1 (2,4; 10,7) per 1.000 patiëntjaren van 2.828 controlepatiënten (mediane behandelingsduur was 5,1 maanden voor met Humira behandelde patiënten en 4,0 maanden voor controlepatiënten). Het percentage (95% betrouwbaarheidsinterval) van niet-melanotische huidkankers was 9,7 (6,6; 14,3) per 1.000 patiëntjaren bij met Humira behandelde patiënten en 5,1 (2,4; 10, 7) per 1.000 patiëntjaren bij controlepatiënten. Van deze huidkankers kwamen plaveiselcelcarcinomen voor met percentages (95% betrouwbaarheidsinterval) van 2,6 (1,2; 5,5) per 1.000 patiëntjaren bij met Humira behandelde patiënten en 0,7 (0,1; 5,2) per 1.000 patiëntjaren bij controlepatiënten. Het percentage lymfomen (95% betrouwbaarheidsinterval) was 0,7 (0,2, 3,0) per 1.000 patiëntjaren bij met Humira behandelde patiënten en 1,5 (0,4; 5,8) per 1.000 patiëntjaren bij controlepatiënten.
Wanneer delen van deze onderzoeken en zowel lopende als voltooide open-label extensieonderzoeken met een gemiddelde duur van ongeveer 3,4 jaar met inbegrip van 5.727 patiënten en meer dan 24.568 patiëntjaren therapie worden gecombineerd, wordt het waargenomen aantal neoplasmata anders dan lymfoom en niet-melanotische huid kanker, is ongeveer 8,8 per 1.000 patiëntjaren. Het waargenomen aantal niet-melanotische huidkanker is ongeveer 10,3 per 1.000 patiëntjaren en het waargenomen aantal lymfomen is ongeveer 1,4 per 1.000 patiëntjaren.
In een postmarketingervaring van januari 2003 tot december 2010, voornamelijk bij patiënten met reumatoïde artritis, is het gerapporteerde aantal neoplasmata ongeveer 2,7 per 1.000 behandelings-/patiëntjaren. De gerapporteerde percentages voor niet-melanotische huidkankers en lymfomen zijn respectievelijk ongeveer 0,2 en 0,3 per 1.000 behandelings-/patiëntjaren (zie rubriek 4.4).
Zeldzame gevallen van hepatosplenisch T-cellymfoom zijn gemeld tijdens postmarketingervaring bij patiënten die werden behandeld met adalimumab (zie rubriek 4.4).
auto-antilichamen
In reumatoïde artritis IV-onderzoeken werden bij verschillende gelegenheden serummonsters van patiënten getest op auto-antilichamen.In deze onderzoeken werden 11,9% van de patiënten behandeld met Humira en 8,1%% van de placebo- en actief-gecontroleerde patiënten die negatieve antinucleaire antilichaamwaarden hadden bij opname hadden positieve waarden in week 24. Twee van de 3.441 patiënten die met Humira werden behandeld bij alle reumatoïde artritis en artritis psoriatica vertoonden klinische symptomen die wezen op het ontstaan van een lupusachtig syndroom. Patiënten verbeterden na stopzetting van de therapie. Geen enkele patiënt ontwikkelde lupus nefritis of centrale symptomen van het zenuwstelsel.
Lever-gal-gebeurtenissen
In gecontroleerde fase 3-klinische onderzoeken met Humira bij patiënten met reumatoïde artritis en artritis psoriatica met een follow-upperiode van 4 tot 104 weken, kwamen ALAT-verhogingen van meer dan of gelijk aan 3 keer de maximale normale waarde voor bij 3,7% van de Humira- behandelde patiënten en 1,6% van de met controle behandelde patiënten.
In gecontroleerde klinische fase 3-onderzoeken met Humira bij patiënten met plaque psoriasis met een controleperiode variërend van 12 tot 24 weken, traden ALAT-verhogingen groter dan of gelijk aan 3 keer de maximale normale waarde op bij "1,8% van de met Humira behandelde patiënten en 1,8 % van de met controle behandelde patiënten.
In gecontroleerde klinische fase 3-onderzoeken met Humira bij patiënten met polyarticulaire juveniele idiopathische artritis van 4 tot 17 jaar en bij patiënten met enthesitis-geassocieerde artritis van 6 tot 17 jaar, ALAT-verhogingen groter dan of gelijk aan een 3-voudige ULN kwam voor bij 6,1% van de met Humira behandelde patiënten en bij 1,3% van de met controle behandelde patiënten. De meeste verhogingen van ALAT-transaminase traden op bij gelijktijdig gebruik van methotrexaat Er waren geen verhogingen van ALAT-transaminase ≥ 3 x ULN in het klinische fase 3-onderzoek van Humira bij patiënten met polyarticulaire juveniele idiopathische artritis.
In gecontroleerde klinische fase 3-onderzoeken met Humira bij patiënten met de ziekte van Crohn en colitis ulcerosa met follow-upperiodes variërend van 4 tot 52 weken, traden ALAT-verhogingen op van meer dan of gelijk aan 3 keer de maximale normale waarde. patiënten en 0,9% van de met controle behandelde patiënten.
In de fase 3-studie van Humira bij pediatrische patiënten met de ziekte van Chron, waarin de veiligheid en werkzaamheid werden beoordeeld van twee op het gewicht aangepaste doseringsschema's voor onderhoudstherapie na een voor het gewicht aangepaste inductietherapie tot 52 weken, werden ALT-spiegels ≥ 3 x ULN gevonden in 2,6% van alle patiënten blootgesteld aan gelijktijdige behandeling met immunosuppressiva bij aanvang.
In klinische onderzoeken waren bij alle indicaties patiënten met verhoogde transaminasespiegels asymptomatisch en in de meeste gevallen waren de verhogingen van voorbijgaande aard en verdwenen ze tijdens de behandeling. Post-marketing gevallen van leverfalen en minder ernstige leveraandoeningen die vooraf kunnen gaan aan leverfalen, zoals hepatitis, waaronder auto-immuunhepatitis, zijn echter ook gemeld bij patiënten die met adalimumab werden behandeld.
Gelijktijdige behandeling met azathioprine/6-mercaptopurine
In onderzoeken naar de ziekte van Crohn bij volwassenen werden hogere incidenties van bijwerkingen die verband houden met ernstige infecties en maligniteiten waargenomen bij de combinatie van Humira en azathioprine/6-mercaptopurine in vergelijking met alleen Humira.
Melding van vermoedelijke bijwerkingen
Het melden van vermoedelijke bijwerkingen die optreden na toelating van het geneesmiddel is belangrijk, omdat het een continue controle van de baten/risicoverhouding van het geneesmiddel mogelijk maakt.Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het Italiaanse Geneesmiddelenbureau. , website: http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
Tijdens klinische onderzoeken werd geen dosisgerelateerde toxiciteit waargenomen. De hoogste geëvalueerde dosis was meerdere doses van 10 mg/kg intraveneus; deze dosis komt overeen met ongeveer 15 maal de aanbevolen dosis.
05.0 FARMACOLOGISCHE EIGENSCHAPPEN
05.1 Farmacodynamische eigenschappen
Farmacotherapeutische categorie: selectieve immunosuppressiva. ATC-code: L04AB04
Werkingsmechanisme
Adalimumab bindt zich selectief aan TNF en neutraliseert de biologische functie ervan door de interactie met de TNF-receptoren van het celmembraan, p55 en p75, te blokkeren.
Adalimumab moduleert ook biologische reacties die worden geïnduceerd of gereguleerd door TNF, waaronder veranderingen in de niveaus van adhesiemoleculen die verantwoordelijk zijn voor leukocytmigratie (ELAM-1, VCAM-1 en ICAM-1 met een IC50 van 0,1-0, 2 nM).
Farmacodynamische effecten
Na behandeling met Humira werd bij patiënten met reumatoïde artritis een snelle afname van de acute fase-eiwitten, ontstekingsindices (C-reactief proteïne -PCR, erytrocytsedimentatiesnelheid -VES) en serumcytokines (IL-6) waargenomen in vergelijking met de basale. Serumspiegels van matrixmetalloproteïnasen (MMP-1 en MMP-3), die betrokken zijn bij weefselremodellering die verantwoordelijk is voor de vernietiging van kraakbeen, waren ook verlaagd na toediening van Humira. Met Humira behandelde patiënten vertoonden over het algemeen een verbetering van de chemie van het bloed, tekenen van chronische ontsteking.
Een snelle afname van de C-reactief proteïne (CRP)-spiegels werd ook waargenomen na behandeling met Humira bij patiënten met polyarticulaire juveniele idiopathische artritis, de ziekte van Crohn en colitis ulcerosa. Bij patiënten met de ziekte van Crohn werd een afname waargenomen in het aantal cellen dat ontstekingsmarkers in de dikke darm tot expressie brengt, waaronder een significante afname van de TNFα-expressie.Endoscopische onderzoeken van het darmslijmvlies hebben genezing van het slijmvlies aangetoond bij patiënten die met adalimumab werden behandeld.
Klinische werkzaamheid en veiligheid
Reumatoïde artritis
Humira is geëvalueerd bij meer dan 3.000 patiënten in alle klinische onderzoeken naar reumatoïde artritis. De werkzaamheid en veiligheid van Humira werden geëvalueerd in vijf gerandomiseerde, dubbelblinde, goed gecontroleerde onderzoeken. Sommige patiënten zijn tot 120 maanden behandeld.
RA-onderzoek I werd uitgevoerd bij 271 patiënten ≥ 18 jaar met matige tot ernstige actieve reumatoïde artritis die ongevoelig waren voor ten minste één DMARD, waaronder methotrexaat, in doses variërend van 12,5 tot 25 mg (10 mg bij intolerantie voor methotrexaat) per week en bij wie de dosis metatrexaat bleef constant op 10-25 mg per week. Humira 20, 40 of 80 mg of placebo werd gedurende 24 weken eenmaal per twee weken gegeven.
Onderzoek AR II onderzocht 544 patiënten ≥ 18 jaar met matige tot ernstige actieve reumatoïde artritis met onvoldoende respons op ten minste één DMARD-geneesmiddel. Doses van 20 of 40 mg Humira werden om de twee weken toegediend via subcutane injectie met placebo om de twee weken, of wekelijks gedurende 26 weken; placebo werd elke week gedurende dezelfde duur gegeven. Het gebruik van andere DMARD's was niet toegestaan.
Onderzoek AR III omvatte 619 patiënten, ≥ 18 jaar oud, met matige tot ernstige actieve reumatoïde artritis die onvoldoende reageerden op methotrexaattherapie in doses variërend van 12,5 tot 25 mg, of intolerantie voor 10 mg methotrexaat elke week. In dit onderzoek werden 3 groepen gevormd. De eerste kreeg 52 weken lang wekelijks placebo-injecties. De tweede kreeg Humira 20 mg per week gedurende 52 weken, terwijl de derde Humira 40 mg om de twee weken en placebo-injecties om de twee weken kreeg. Na voltooiing van de eerste 52 weken werden 457 patiënten geïncludeerd in een open-label verlengingsfase waarin Humira/MTX werd toegediend in een dosis van 40 mg eenmaal per twee weken gedurende maximaal 10 jaar.
In het AR IV-onderzoek werd eerst de veiligheid van Humira beoordeeld bij 636 patiënten met matige tot ernstige actieve reumatoïde artritis in de leeftijd ≥ 18 jaar. De onderzoekspopulatie bestond uit zowel patiënten die nooit met DMARD's werden behandeld als patiënten die een reeds bestaande antireumatische therapie hadden voortgezet, op voorwaarde dat deze gedurende minimaal 28 dagen stabiel was. Deze therapieën omvatten methotrexaat, leflunomide, hydroxychloroquine, sulfasalazine en/of goudzouten.Patiënten werden gerandomiseerd om gedurende 24 weken elke twee weken Humira 40 mg of placebo te krijgen.
In het AR-onderzoek V werden 799 volwassen patiënten geëvalueerd die nooit eerder met methotrexaat waren behandeld en die matige tot ernstige vroege actieve reumatoïde artritis hadden (gemiddelde duur van de ziekte minder dan 9 maanden). Deze studie evalueerde de werkzaamheid van Humira 40 mg eenmaal per twee weken gegeven in combinatietherapie met methotrexaat, Humira 40 mg gegeven als monotherapie eenmaal per twee weken en methotrexaat alleen bij het verminderen van de tekenen en symptomen van ziekte en de index van progressie van gewrichtsschade veroorzaakt door reumatoïde artritis gedurende 104 weken.
Het primaire eindpunt van AR-onderzoeken I, II, III en de secundaire eindpunten van AR IV was het bepalen van het percentage patiënten dat een ACR 20-respons bereikte in week 24 of 26. Het primaire doel van het AR V-onderzoek was het evalueren van de percentage patiënten dat een ACR 50-respons bereikte in week 52. Daarnaast hadden de AR-onderzoeken III en V als hoofddoel het aantonen van remming van ziekteprogressie (door middel van röntgenonderzoek) in week 52. Het AR-onderzoek III had ook als hoofddoel verbetering van de kwaliteit van leven aan te tonen.
ACR-reactie
De percentages van met Humira behandelde patiënten die een ACR-respons van 20, 50 en 70 bereikten, waren vergelijkbaar in AR-onderzoeken I, II en III. De resultaten voor behandeling met 40 mg om de twee weken zijn samengevat in Tabel 3.
In RA-onderzoeken I-IV werden alle parameters geëvalueerd voor de definitie van de ACR-respons (aantal pijnlijke en gezwollen gewrichten, evaluatie van ziekteactiviteit door de arts en patiënt, evaluatie van pijn door de patiënt, invaliditeitsindex - HAQ) en CRP-waarden (mg/dL) verbeterde na 24 of 26 weken in vergelijking met placebo. In studie AR III hielden deze verbeteringen gedurende 52 weken aan.
In de open-label verlengingsfase van het AR III-onderzoek behielden de meeste patiënten die een ACR-respons ondervonden, de respons wanneer ze de behandeling gedurende 10 jaar voortzetten. Van de in totaal 207 patiënten die werden gerandomiseerd naar Humira 40 mg eenmaal per twee weken, gingen 114 gedurende 5 jaar door met Humira 40 mg eenmaal per twee weken. Hiervan hadden 86 patiënten (75,4%) een ACR 20-respons; 72 patiënten (63,2%) hadden een ACR 50-respons; en 41 patiënten (36%) hadden een respons op ACR 70. Van de in totaal 207 patiënten zetten 81 de behandeling voort met Humira 40 mg eenmaal per twee weken gedurende 10 jaar. Hiervan hadden 64 patiënten (79,0%) een ACR 20-respons; 56 patiënten (69,1%) hadden een ACR 50-respons; en 43 patiënten (53,1%) hadden een ACR 70-respons.
In RA-onderzoek IV was de ACR 20-respons van met Humira behandelde patiënten in combinatie met conventionele therapie statistical statistisch significant beter dan bij patiënten behandeld met placebo in combinatie met traditionele geneesmiddelen (p
In RA-onderzoeken I-IV bereikten patiënten die met Humira werden behandeld al 1-2 weken na aanvang van de behandeling statistisch significant hogere ACR 20- en 50-responsen dan placebo.
In RA-onderzoek V resulteerde Humira/methotrexaat-combinatietherapie bij patiënten met vroege reumatoïde artritis die nooit eerder met methotrexaat waren behandeld, in snellere en significant grotere ACR-responsen dan methotrexaat monotherapie en Humira monotherapie in week 52 en deze responsen hielden aan gedurende 104 weken ( zie Tabel 4).
In week 52 bereikte 42,9% van de patiënten die de combinatietherapie Humira/methotrexaat kregen klinische remissie (DAS28
radiologische reactie
In het AR-onderzoek III, waarbij met Humira behandelde patiënten een gemiddelde ziekteduur van ongeveer 11 jaar hadden, werd structurele schade radiografisch beoordeeld en uitgedrukt als een verandering in de gewijzigde Total Sharp Score (TSS) en gerelateerde componenten, erosie en vernauwing van de gewrichtsspleet ( JSN)-indexen.Humira/MTX-behandelde patiënten vertoonden significant minder radiologische progressie dan patiënten die alleen MTX kregen, na 6 en 12 maanden (zie Tabel 5).
In de open-label uitbreiding van AR-onderzoek III blijft de vermindering van de progressie van structurele schade gedurende 8 en 10 jaar behouden bij een subgroep van patiënten.Na 8 jaar werden 81 van de 207 patiënten die oorspronkelijk werden behandeld met Humira 40 mg elke andere weken werden radiologisch beoordeeld na 5 jaar. Onder deze 48 patiënten vertoonden geen progressie van structurele schade gedefinieerd door een mTSS-verandering van 0,5 of minder vanaf baseline. Na 10 jaar werden 79 van de 207 patiënten die oorspronkelijk werden behandeld met Humira 40 mg eenmaal per twee weken week werd radiologisch geëvalueerd. Van deze patiënten vertoonden 40 patiënten geen progressie van structurele schade gedefinieerd door een verandering in mTSS van 0,5 of minder vanaf baseline.
naar methotrexaat
b 95% betrouwbaarheidsinterval voor verschillen in indexveranderingen tussen methotrexaat en Humira.
c Gebaseerd op ranganalyse
dJoint Space Narrowing (verkleining van de voegspleet)
In de AR-studie V werd structurele gewrichtsschade radiografisch beoordeeld en uitgedrukt in termen van de verandering in de gewijzigde Total Sharp Score (zie tabel 6).
Na 52 weken en 104 weken behandeling was het percentage patiënten zonder progressie (verandering t.o.v. baseline in gewijzigde Total Sharp Score & ≤ 0,5) significant hoger met Humira/methotrexaat combinatietherapie (respectievelijk 63,8% en 61,2%) vergeleken met methotrexaat monotherapie (respectievelijk 37,4% en 33,5%, p
Kwaliteit van leven en fysiek functioneren
De kwaliteit van leven en fysiek functioneren werden beoordeeld met de invaliditeitsindex verkregen via de Health Assessment Questionnaire (HAQ), in vier originele, adequate en goed gecontroleerde onderzoeken, en was een van de primaire eindpunten van AR-onderzoek III in week 52. Alle Humira regimes in de vier onderzoeken lieten statistisch significante verbeteringen zien in de invaliditeitsindex van de placebo-HAQ tussen baseline en maand 6 in vergelijking met placebo en in het AR-onderzoek III werd hetzelfde resultaat waargenomen in week 52. De analyse van de algemene gezondheidstoestand, beoordeeld door de Short Form Health Survey (SF -36) in de vier onderzoeken, ondersteunt deze conclusies voor alle Humira-doseringsregimes met statistisch gerapporteerde resultaten significant wat betreft de indices van fysieke activiteit, pijn en welzijn, geregistreerd met Humira 40 mg op zich keer afwisselend. Een statistisch significante afname van het gevoel van vermoeidheid, zoals blijkt uit de indices van de functionele evaluatie met betrekking tot de behandeling van chronische ziekten (FACIT) werd gevonden in alle drie de onderzoeken waarin het werd geëvalueerd (onderzoeken AR I, III, IV).
In het AR III-onderzoek behield de meerderheid van de proefpersonen die verbetering in fysiek functioneren bereikten en die de behandeling voortzetten, verbetering gedurende 520 weken (120 maanden) open-label behandeling. De verbetering van de kwaliteit van leven werd gemeten tot week 156 (36 maanden) en de verbetering hield in de tijd aan.
In het AR-onderzoek V vertoonden de invaliditeitsindex zoals beoordeeld door HAQ en de fysieke component van SF 36 superieure verbetering (p
Juveniele idiopathische artritis
Polyarticulaire juveniele idiopathische artritis (pJIA)
De veiligheid en werkzaamheid van Humira zijn beoordeeld in twee onderzoeken (pJIA I en II) bij kinderen met actieve polyarticulaire of polyarticulaire juveniele idiopathische artritis, die verschillende typen JIA hadden (meestal reumafactor-negatieve of positieve polyartritis en uitgebreide oligoartritis).
pJIA I
De veiligheid en werkzaamheid van Humira werden beoordeeld in een multicenter, gerandomiseerd, dubbelblind onderzoek met parallelle groepen bij 171 kinderen (in de leeftijd van 4-17 jaar) met polyarticulaire juveniele idiopathische artritis (JIA). = OL LI, patiënten werden gestratificeerd in twee groepen, de MTX (methotrexaat) groep en de MTX onbehandelde groep. De arm die niet met MTX werd behandeld, was nog nooit eerder met MTX behandeld of was ten minste twee weken voorafgaand aan de toediening van het onderzoeksgeneesmiddel gestopt met het gebruik van MTX. Patiënten kregen constante doses niet-steroïde anti-inflammatoire geneesmiddelen (NSAID's) en/of prednison (≤0,2 mg/kg/dag of maximaal 10 mg/dag). Tijdens de OL LI-fase kregen alle patiënten Humira 24 mg/dag. m2 tot een maximale dosis van 40 mg eenmaal per twee weken gedurende 16 weken De verdeling van patiënten naar leeftijd en de minimale, gemiddelde en maximale dosis die tijdens de OL LI-fase is toegediend, worden weergegeven in Tabel 7.
Tabel 7
Verdeling van patiënten naar leeftijd en dosis adalimumab toegediend tijdens de OL LI-fase
Patiënten die in week 16 een pediatrische ACR30-respons vertoonden, kwamen in aanmerking voor randomisatie in de dubbelblinde (DB)-fase van het onderzoek en kregen Humira 24 mg/m2 tot een maximum van 40 mg of placebo om de twee weken voor nog eens 32 weken of totdat de ziekte opflakkert. De criteria voor het definiëren van de verergering van de ziekte werden gedefinieerd op basis van een verslechtering groter dan of gelijk aan 30% (≥ 30%) vergeleken met de uitgangswaarde van 3 of meer van de 6 hoofdcriteria van de "ACR Pediatric Core", in de aanwezigheid van 2 of meer actieve gewrichten, en gebaseerd op een verbetering van meer dan 30% in niet meer dan 1 van de bovengenoemde 6. Na 32 weken of wanneer de ziekte opvlamde, werden patiënten geacht de noodzakelijke vereisten te hebben om te worden toegelaten tot de open-label verlengingsfase.
Tabel 8
PedACR30-respons tijdens het JIA-onderzoek
a Ped ACR 30/50/70-responsen in week 48 waren significant groter dan die bereikt bij met placebo behandelde patiënten
bp = 0,015
cp = 0,031
Van degenen die in week 16 reageerden op de behandeling (n = 144), bleven de Ped ACR 30/50/70/90-responsen gedurende maximaal zes jaar behouden tijdens de OLE-fase bij patiënten die Humira kregen gedurende de hele studio. In totaal werden 19 proefpersonen, waaronder 11 uit de kerngroep van 4 tot 12 jaar en 8 uit de kernleeftijdsgroep van 13 tot 17 jaar, gedurende 6 jaar of langer behandeld.
De algehele respons was over het algemeen beter en minder patiënten ontwikkelden antilichamen bij behandeling met de combinatie Humira en MTX in vergelijking met alleen toegediende Humira. Rekening houdend met deze resultaten wordt het gebruik van Humira aanbevolen in combinatie met MTX en als monotherapie bij patiënten voor wie het gebruik van MTX niet wordt aanbevolen (zie rubriek 4.2).
pJIA II
De veiligheid en werkzaamheid van Humira zijn geëvalueerd in een open-label multicenter onderzoek bij 32 kinderen (2-2 lichaamsoppervlak Humira tot een maximum van 20 mg eenmaal per twee weken als een enkelvoudige subcutane dosis gedurende ten minste 24 weken. in het onderzoek gebruikten de meeste proefpersonen gelijktijdig MTX, waarbij sommige proefpersonen melding maakten van het gebruik van corticosteroïden of niet-steroïde anti-inflammatoire geneesmiddelen (NSAID's).
In week 12 en week 24 was de PedACR30-respons respectievelijk 93,5% en 90,0% volgens de geobserveerde gegevensbenadering. De proporties proefpersonen met PedACR50/70/90 in week 12 en week 24 waren respectievelijk 90,3% / 61,3% / 38,7% en 83,3% / 73,3% / 36,7% Onder degenen die reageerden (PedACR30) in week 24 (n = 27 van de 30 patiënten), bleef de PedACR30-respons gedurende maximaal 60 weken behouden bij patiënten die Humira kregen gedurende deze periode in de open lucht -label verlengingsonderzoek. In totaal werden ≤ 20 proefpersonen gedurende 60 weken of langer behandeld.
Artritis geassocieerd met enthesitis
De veiligheid en werkzaamheid van Humira werden geëvalueerd in een multicenter, gerandomiseerd, dubbelblind onderzoek bij 46 pediatrische patiënten (6 tot 17 jaar) met matige artritis-geassocieerde enthesitis. Patiënten werden gerandomiseerd om Humira 24 mg/m2 lichaamsoppervlak te krijgen. gebied, tot een maximum van 40 mg, of placebo eenmaal per twee weken gedurende 12 weken.De dubbelblinde periode werd gevolgd door een open-label onderzoeksperiode, waarin patiënten Humira 24 mg/m2 lichaamsoppervlak kregen, tot een maximaal 40 mg subcutaan eenmaal per twee weken, gedurende nog eens 192 weken Het primaire eindpunt was de procentuele verandering in het aantal actieve gewrichten met artritis vanaf baseline tot week 12 (zwelling niet door misvorming of gewrichten met bewegingsverlies plus pijn en/of pijn), wat werd bereikt met een gemiddelde procentuele afname van -62,6% (gemiddelde procentuele verandering - 88,9%) bij patiënten in de Humira-groep vergeleken met -11,6% (gemiddelde procentuele verandering - 50,0%) bij patiënten in de placebogroep. De verbetering in het aantal actieve gewrichten met artritis hield aan tijdens de open-label periode van het onderzoek tot week 52. Hoewel niet statistisch significant, vertoonden de meeste patiënten klinische verbetering in het secundaire eindpunt, zoals het aantal plaatsen van enthesitis, pijnlijke aantal gewrichten (TJC), aantal gezwollen gewrichten (SJC), ACR 50-respons bij kinderen en ACR 70-respons bij kinderen.
Axiale spondyloartritis
Spondylitis ankylopoetica (AS)
De toediening van Humira 40 mg eenmaal per twee weken door 393 patiënten werd geëvalueerd in twee 24 weken durende gerandomiseerde dubbelblinde placebogecontroleerde onderzoeken bij proefpersonen met actieve spondylitis ankylopoetica (waarbij de gemiddelde baselinescore van "ziekteactiviteit [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)] was gelijk aan 6,3 in alle geanalyseerde groepen) die een ontoereikende respons ontwikkelden op conventionele therapie. Negenenzeventig patiënten (20,1%) werden behandeld met gelijktijdige therapie met DMARD's en 37 patiënten (9,4%) met glucocorticoïden. geblindeerde periode werd gevolgd door een open-label periode waarin patiënten Humira 40 mg eenmaal per twee weken subcutaan kregen gedurende een extra periode tot 28 weken. 12, of week 16 of week 20, werden gegeven gebruikten subcutaan 40 mg adalimumab als vroege open-label noodtherapie om de twee weken en werden vervolgens behandeld als non-responders in dubbelblinde statistische analyses.
In een groter AS I-onderzoek waarin 315 patiënten werden geanalyseerd, lieten de resultaten een statistisch significante verbetering zien van de tekenen en symptomen van spondylitis ankylopoetica bij met Humira behandelde patiënten in vergelijking met met placebo behandelde patiënten.
De significante respons werd voor het eerst waargenomen in week 2 en hield aan gedurende een periode van 24 weken (Tabel 9).
Tabel 9
Werkzaamheidsreacties in placebo-gecontroleerde spondylitis ankylopoetica - onderzoek I Vermindering van tekenen en symptomen
***, ** Statistisch significant op p
a Beoordelingen van spondylitis ankylopoetica
bBath Spondylitis ankylopoetica Activiteitsindex
De met Humira behandelde patiënten ondervonden een significant hogere verbetering in week 12, die werd gehandhaafd gedurende de duur van de behandeling tot en met week 24 in zowel de SF36 als de Ankylosing Spondylitis Quality of Life Questionnaire (ASAQoL).
Vergelijkbare trends (niet allemaal statistisch significant) werden waargenomen in een kleinere, gerandomiseerde, dubbelblinde, placebogecontroleerde AS II-studie bij 82 volwassen patiënten met actieve spondylitis ankylopoetica.
Axiale spondyloartritis zonder radiografisch bewijs van SA
Humira, gegeven in een dosis van 40 mg eenmaal per twee weken, werd geëvalueerd bij 185 patiënten met actieve niet-radiografische axiale spondyloartritis in een gerandomiseerde, 12 weken durende, dubbelblinde, placebogecontroleerde studie (de gemiddelde uitgangswaarde van ziekteactiviteit [ Bath Ankylosing Spondylitis Disease Activity Index (BASDAI)] was 6,4 voor met Humira behandelde patiënten en 6,5 voor met placebo behandelde patiënten) die onvoldoende reageerden op of intolerant waren voor een of meer NSAID's, of een contra-indicatie voor NSAID's hadden.
Drieëndertig patiënten (18%) werden gelijktijdig behandeld met ziektemodificerende antireumatische geneesmiddelen en 146 patiënten (79%) met NSAID's bij aanvang. De dubbelblinde periode werd gevolgd door een open-label periode waarin patiënten Humira 40 mg eenmaal per twee weken subcutaan kregen gedurende maximaal 144 weken. De resultaten van week 12 lieten een statistisch significante verbetering zien van de tekenen en symptomen van actieve niet-radiografische axiale spondyloartritis bij patiënten die met Humira werden behandeld in vergelijking met placebo (tabel 10).
Tabel 10
Werkzaamheidsrespons in placebogecontroleerd onderzoek bij axiale spondyloartritis - vermindering van tekenen en symptomen
Gezondheidsgerelateerde kwaliteit van leven en fysiek functioneren werden beoordeeld met behulp van de HAQ-S- en SF-36-vragenlijsten. Humira vertoonde een statistisch significant grotere verbetering ten opzichte van baseline dan placebo in de totale HAQ-S-score en SF-36 Physical Component Score in week 12.
Psoriatische arthritis
Humira, gegeven in een dosis van 40 mg eenmaal per twee weken, is onderzocht bij patiënten met matige tot ernstige actieve artritis psoriatica in twee placebogecontroleerde onderzoeken, PsA I en II. Tijdens de 24 weken durende PsA I-studie werden 313 volwassen patiënten behandeld die onvoldoende reageerden op niet-steroïde anti-inflammatoire medicamenteuze behandeling en van hen nam ongeveer 50% methotrexaat. In de 12 weken durende PsA II-studie werden 100 patiënten behandeld die onvoldoende reageerden op de DMARD-therapie. Aan het einde van beide onderzoeken namen 383 patiënten deel aan een open-label extensieonderzoek en kregen ze om de twee weken Humira 40 mg.
Vanwege het beperkte aantal onderzochte patiënten is er onvoldoende bewijs voor de werkzaamheid van Humira bij patiënten met spondylitis ankylopoetica-achtige artritis psoriatica.
Tabel 11
ACR-responsen in placebogecontroleerde onderzoeken bij gevallen van artritis psoriatica (percentage patiënten)
ACR-responsen in PsA I waren vergelijkbaar met en zonder gelijktijdige behandeling met methotrexaat.
ACR-responsen in het open-label verlengingsonderzoek bleven tot 136 weken behouden.
Radiologische veranderingen werden geëvalueerd in de onderzoeken naar artritis psoriatica Röntgenfoto's van de handen, polsen en voeten werden genomen bij aanvang en in week 24, tijdens de dubbelblinde fase wanneer patiënten werden behandeld met Humira of placebo, en in week 48, wanneer alle patiënten werden behandeld met open-label Humira.Modified Total Sharp Score (mTSS) werd gebruikt met inbegrip van distale interfalangeale gewrichten (dwz verschillend van Total Sharp Score gebruikt voor reumatoïde artritis).
Behandeling met Humira verminderde in vergelijking met placebobehandeling de snelheid van progressie van perifere gewrichtsschade zoals gemeten door veranderingen in gewijzigde Total Sharp Score vanaf baseline (gemiddelde ± SD) 0,8 ± 2,5 in placebogroep (in week 24) versus 0,0 ± 1,9 (p
Bij met Humira behandelde patiënten zonder progressie van radiologische schade vanaf baseline tot week 48 (n = 102), bleef 84% geen progressie van radiologische schade vertonen gedurende 144 weken behandeling.
Met Humira behandelde patiënten vertoonden een statistisch significante verbetering in fysiek functioneren in week 24 in vergelijking met met placebo behandelde patiënten, zoals beoordeeld door de HAQ en Short Form Health Survey (SF 36). Verbetering in fysiek functioneren hield aan tot week 136 in het open-label verlengingsonderzoek .
Psoriasis
De veiligheid en werkzaamheid van Humira zijn onderzocht bij volwassen patiënten met chronische plaque psoriasis (BSA ≥10% en Psoriasis Area and Severity Index (PASI) ≥12 of ≥10) die in aanmerking kwamen voor systemische therapie of fototherapie, tijdens gerandomiseerde dubbelblinde 73% van de patiënten die waren opgenomen in de psoriasisonderzoeken I en II hadden eerder systemische of fototherapietherapie gekregen De veiligheid en werkzaamheid van Humira werden ook onderzocht bij volwassen patiënten met chronische psoriasis tot matige tot ernstige plaque met gelijktijdig gelokaliseerde hand en/of voet psoriasis, die in aanmerking kwamen voor systemische therapie in een gerandomiseerde dubbelblinde studie (fase III psoriasisstudie).
Psoriasisonderzoek I (REVEAL) evalueerde 1.212 patiënten binnen drie behandelingsperioden. Tijdens periode A kregen de patiënten ofwel placebo ofwel Humira in een startdosis van 80 mg, gevolgd door een dosis van 40 mg om de twee weken, beginnend in de week na de aanvangsdosis. ten minste één PASI 75-respons (waarvan de PASI-score ten minste 75% verbeterde ten opzichte van de uitgangswaarde) werd opgenomen in periode B en ontving een dosis Humira gelijk aan 40 mg, om de twee weken, open label. week 33 en die aanvankelijk gerandomiseerd waren naar actieve therapie tijdens periode A, werden opnieuw gerandomiseerd tijdens periode C om gedurende 19 extra weken 40 mg Humira of placebo te krijgen. was 18,9 en de score van de arts was "s Global Assessment (PGA) was" matig "in 53% van de opgenomen proefpersonen", ernstig "in 41% en" zeer ernstig "in 6,6%.
Psoriasisonderzoek II (CHAMPION) vergeleek de werkzaamheid en veiligheid van Humira met methotrexaat en placebo bij 271 patiënten. Over een periode van 16 weken kregen de patiënten placebo of methotrexaat met een startdosis van 7, 5 mg, daarna verhoogd tot week 12 en met een maximum van 25 mg, of Humira in de startdosis van 80 mg gevolgd door de 40 mg dosis om de twee weken (vanaf de week na de initiële dosis). Er zijn geen gegevens beschikbaar waarin Humira en methotrexaat worden vergeleken na een behandeling van meer dan 16 weken. Bij methotrexaatbehandelde patiënten die een ≥PASI 50-respons bereikten in week 8 en/of week 12, werden geen dosisverhogingen doorgevoerd. In alle behandelingsgroepen was de gemiddelde PASI-score bij aanvang 19,7 en de PGA-score bij aanvang was "mild" (
Patiënten die deelnamen aan alle fase 2- en fase 3-onderzoeken naar psoriasis kwamen in aanmerking voor deelname aan een open-label verlengingsonderzoek, waarbij Humira gedurende een aanvullende periode van ten minste 108 weken werd toegediend.
In psoriasisonderzoeken I en II was het primaire eindpunt het percentage patiënten dat een PASI 75-respons bereikte bij baseline in week 16 (zie tabellen 12 en 13).
In Psoriasis-onderzoek I werd 28% van de patiënten gerandomiseerd naar placebo in week 33 na het bereiken van een PASI 75-respons, vergeleken met 5% van de patiënten die de Humira-therapie voortzetten, p
In Psoriasis-onderzoek I gingen in totaal 233 patiënten, inclusief degenen die een PASI 75-respons bereikten in week 16 en week 33 en die de Humira-therapie voortzetten tot en met week 52, door met Humira in het onderzoek. Bij deze patiënten waren de PASI 75- en PGA-responspercentages die overeenkomen met totale ziekteremissie of minimale ziektepersistentie respectievelijk 74,7% en 59,0% na nog eens 108 weken open-label therapie (voor een totaal van 160 weken continue therapie) . In een analyse uitgevoerd bij alle patiënten die de studie hadden stopgezet vanwege bijwerkingen of gebrek aan werkzaamheid, of die de dosering hadden verhoogd en om deze redenen werden beschouwd als niet reagerend op de therapie, waren de PASI 75-responspercentages en PGA overeenkomende totale ziekteremissie of minimale ziektepersistentie waren respectievelijk 69,6% en 55,7% na nog eens 108 weken open-label therapie (voor een totaal van 160 weken continue therapie).
In totaal 347 patiënten, gekenmerkt door een stabiele respons op de therapie, namen deel aan een open-label vervolgonderzoek om de effecten van het stopzetten en hervatten van de behandeling te evalueren. Tijdens de ontwenningsperiode kwamen de psoriasissymptomen progressief terug met een mediane tijd tot terugval (overgaand tot een "matig" of erger PGA-stadium) van ongeveer 5 maanden. Geen van deze patiënten kreeg reboundverschijnselen tijdens de periode van stopzetting van de behandeling. In totaal bereikte 76,5% van de patiënten (218/285) die de herbehandelingsfase ingingen een PGA-respons die overeenkwam met totale remissie van de ziekte of minimale ziekte na zestien weken therapie, ongeacht of ze al dan niet opflakkeringen van de ziekte hadden gehad tijdens de ontwenningsperiode van het geneesmiddel (respectievelijk 69,1% [123/178] en 88,8% [95/107] van de patiënten die wel of geen opflakkering van de ziekte hadden tijdens de ontwenningsperiode).
Tijdens de hervatting van de behandeling werd een veiligheidsprofiel waargenomen dat zeer vergelijkbaar was met het veiligheidsprofiel dat werd waargenomen tijdens de periode voorafgaand aan het staken van de behandeling.
Aanzienlijke verbeteringen in Dermatology Life Quality Index (DLQI) werden aangetoond in week 16 vanaf baseline in vergelijking met placebo (onderzoeken I en II) en methotrexaat (onderzoek II). In onderzoek I waren verbeteringen in de algehele fysieke en mentale componentscores van SF-36 ook significant in vergelijking met placebo.
In een open-label verlengingsonderzoek bij patiënten die vanwege een PASI-respons van minder dan 50% dosisverhogingen kregen van 40 mg eenmaal per twee weken tot 40 mg eenmaal per week, beoordeeld in week 12 na dosisverhoging, 93 van 349 (26,6% ) bereikte een PASI 75-respons.
Het fase III-psoriasisonderzoek (REACH) vergeleek de werkzaamheid en veiligheid van Humira versus placebo bij 71 patiënten met matige tot ernstige chronische plaque psoriasis en gelokaliseerde hand- en/of voetpsoriasis die een startdosis van 80 mg Humira kregen gevolgd door 40 mg toegediend om de week (vanaf de week na de startdosis) of placebo gedurende 16 weken. In week 16 bereikte een statistisch groter deel van de patiënten die Humira kregen een PGA-respons die overeenkomt met totale of bijna totale remissie van hand- en/of voetziekte vergeleken met patiënten die placebo kregen (respectievelijk 30,6%) versus 4,3%, [P = 0,014]).
ziekte van Crohn
De veiligheid en werkzaamheid van Humira zijn geëvalueerd bij meer dan 1500 patiënten met matig tot ernstig actieve ziekte van Crohn (Crohn's Disease Activity Index (CDAI) 220 en ≤ 450 ) in gerandomiseerde, dubbelblinde, placebogecontroleerde onderzoeken. Gelijktijdige toediening van constante doses aminosalicylaten, corticosteroïden en/of immunomodulerende middelen was toegestaan en 80% van de patiënten bleef ten minste één van deze geneesmiddelen gebruiken.
De inductie van klinische remissie (gedefinieerd als CDAI
Handhaving van klinische remissie werd geëvalueerd in de CD III-studie (CHARM).In CD-onderzoek III kregen 854 patiënten open-label Humira 80 mg in week 0 en 40 mg in week 2. In week 4 werden patiënten gerandomiseerd naar 40 mg eenmaal per twee weken, 40 mg eenmaal per week of placebo. de totale duur van het onderzoek was 56 weken. Patiënten die in week 4 een adequate klinische respons vertoonden (CDAI-daling ≥ 70) werden gestratificeerd en afzonderlijk geanalyseerd van degenen die geen adequate klinische respons vertoonden in week 4. Een geleidelijke verlaging van de dosis corticosteroïden na week 8.
De inductiepercentages van klinische remissie en respons van CD-onderzoek I en CD-onderzoek II worden weergegeven in Tabel 14.
Vergelijkbare remissiepercentages werden waargenomen in de inductiedosisgroep van 160/80 mg en 80/40 mg in week 8 en bijwerkingen kwamen vaker voor in de 160/80 mg dosisgroep.
In CD-onderzoek III ervoer 58% (499/854) van de patiënten in week 4 een adequate klinische respons en werd geëvalueerd in de primaire analyse. Van de patiënten die een adequate klinische respons ervoeren in week 4, was 48% eerder blootgesteld therapie met andere TNF-antagonisten De percentages voor handhaving van remissie en klinische respons zijn weergegeven in Tabel 15. De resultaten voor klinische remissie bleven relatief constant ongeacht eerdere blootstelling aan geneesmiddelen tegen geneesmiddelen.
In week 56 waren ziektegerelateerde ziekenhuisopnames en operaties statistisch significant verminderd met adalimumab in vergelijking met placebo.
Van de patiënten die in week 4 geen adequate respons vertoonden, ervoer 43% van de patiënten die werden behandeld met Humira-onderhoudstherapie een adequate respons in week 12, vergeleken met 30% van de met placebo behandelde patiënten. Deze resultaten suggereren dat sommige patiënten die in week 4 geen adequate respons vertoonden, baat hebben bij voortzetting van de onderhoudstherapie tot en met week 12. Behandeling die langer dan 12 weken duurde, leidde niet tot een significant hoger aantal responsen (zie rubriek 4.2).
117/276 patiënten uit CD-onderzoek I en 272/777 patiënten uit CD-onderzoeken II en III werden gedurende ten minste 3 jaar open-label adalimumab-therapie gevolgd. Bij respectievelijk 88 en 189 patiënten bleef de klinische remissie behouden. De klinische respons (CR-100) bleef behouden bij respectievelijk 102 en 233 patiënten.
Levenskwaliteit
In CD I- en CD II-onderzoeken werd een statistisch significante verbetering van de totale score 4 van de ziektespecifieke inflammatoire darmziekte (IBDQ) bereikt in week 4 bij patiënten die waren gerandomiseerd naar Humira 80/40 mg en 160/80 mg vergeleken met placebo en werd gezien in week 26 en 56 in CD-onderzoek III en tussen de Humira-behandelingsgroepen in vergelijking met de placebogroep.
Ziekte van Crohn bij pediatrische patiënten
Humira is getest in een multicenter, gerandomiseerd, dubbelblind klinisch onderzoek dat was opgezet om de werkzaamheid en veiligheid te evalueren van gewichtsafhankelijke dosisafhankelijke inductie- en onderhoudsbehandeling (matige tot ernstige ziekte van Crohn (CD). gedefinieerd door een Pediatric Crohn's Disease Activity Index (PCDAI)-score van > 30. De proefpersonen moeten hebben gefaald bij conventionele therapie (inclusief een corticosteroïd en/of immunomodulator) voor CD.Bovendien kunnen proefpersonen eerder de respons verloren hebben of intolerant waren voor infliximab.
Alle proefpersonen kregen open-label inductietherapie met een dosis gebaseerd op hun lichaamsgewicht bij baseline: 160 mg in week 0 en 80 mg in week 2 voor proefpersonen die 40 kg wogen, en respectievelijk 80 mg en 40 mg voor proefpersonen met gewicht
In week 4 werden de proefpersonen, op basis van hun lichaamsgewicht, 1:1 gerandomiseerd naar het onderhoudsschema met lage dosis of standaarddosis, zoals weergegeven in tabel 16.
Effectiviteit resultaten
Het primaire eindpunt van het onderzoek was klinische remissie in week 26, gedefinieerd door een PCDAI-score & 10.
Klinische remissie- en klinische responspercentages (gedefinieerd als verlaging van de PCDAI-score van ten minste 15 punten ten opzichte van de uitgangswaarde) worden weergegeven in Tabel 17. De percentages voor stopzetting van corticosteroïden of immunomodulatoren worden weergegeven in Tabel 18.
Statistisch significante stijgingen (verbeteringen) in Body Mass Index en lengtesnelheid vanaf baseline tot week 26 en week 52 werden waargenomen voor beide behandelingsgroepen.
Statistisch en klinisch significante verbeteringen ten opzichte van baseline in parameters voor kwaliteit van leven (inclusief IMPACT III) werden ook waargenomen in beide behandelingsgroepen.
Colitis ulcerosa
De veiligheid en werkzaamheid van meervoudige doses Humira werden geëvalueerd bij volwassen patiënten met matige tot ernstige actieve colitis ulcerosa (Mayo-score 6 tot 12 met een endoscopische subscore van 2 tot 3) in gerandomiseerde, dubbelblinde onderzoeken, placebogecontroleerd.
In onderzoek UC-I werden 390 patiënten die niet eerder met TNF-antagonisten waren behandeld, gerandomiseerd naar placebo in week 0 en 2, 160 mg Humira in week 0 gevolgd door 80 mg in week 2 of 80 mg in week 0 gevolgd. in week 2. Na week 2 kregen de patiënten in beide adalimumab-armen 40 mg eenmaal per twee weken. Klinische remissie (gedefinieerd als Mayo-score ≤ 2 zonder subscores> 1) werd beoordeeld in week 8.
In onderzoek UC-II namen 248 patiënten Humira 160 mg in week 0, 80 mg in week 2 en 40 mg eenmaal per twee weken, en 246 patiënten namen placebo. Inductie van remissie werd beoordeeld in week 8 en handhaving van remissie in week 52.
In onderzoek UC-I (respectievelijk 18% vs. 9%, p = 0,031) en in onderzoek UC-II (respectievelijk 17% vs. 9%, p = 0,019) bereikten de met Humira 160/80 mg geïnduceerde patiënten klinische remissie vergeleken met placebo in week 8 in statistisch significant hogere percentages. In onderzoek UC-II waren onder met Humira behandelde patiënten in remissie in week 8, 21/41 (51%) in remissie in week 52.
De algemene resultaten van het UC-II-populatieonderzoek worden weergegeven in Tabel 19.
Tabel 19
Mucosale respons, remissie en genezing in onderzoek UC-II (percentage patiënten)
Van de patiënten die in week 8 reageerden, vertoonde 47% klinische respons, 29% was in remissie, 41% had mucosale genezing en 20% was in steroïde-vrije klinische remissie gedurende ≥ 90 dagen in week 52.
Bij ongeveer 40% van de patiënten die deelnamen aan UC-II had een eerdere anti-TNF-behandeling met infliximab gefaald. De werkzaamheid van adalimumab bij die patiënten was verminderd in vergelijking met die getoond bij patiënten die niet eerder met anti-TNF waren behandeld.Van de patiënten bij wie eerdere anti-TNF-behandeling had gefaald, bereikte 3% van de placebo-arm en 10% van de adalimumab-arm remissie op week 52.
Patiënten die deelnamen aan UC-I en UC-II kregen de mogelijkheid om deel te nemen aan een uitgebreide open langetermijnstudie (UC-III). Na drie jaar therapie op basis van adalimumab was 75% (301/402) nog steeds in klinische remissie volgens de partiële Mayo-score.
Ziekenhuisopname tarief
Tijdens de 52 weken van onderzoeken UC-I en UC-II werd een afname van ziekenhuisopnames door alle oorzaken en colitis ulcerosa-gerelateerde ziekenhuisopnames waargenomen in de adalimumabgroep vergeleken met de placebogroep. Het aantal ziekenhuisopnames door alle oorzaken was 0,18 proefpersonen per jaar in de adalimumab-behandelingsgroep vergeleken met de placebogroep (0,26 proefpersonen per jaar) en de corresponderende gegevens over ziekenhuisopnames gerelateerd aan colitis ulcerosa waren 0,12 proefpersonen per jaar, vergeleken met 0,22 proefpersonen per jaar.
Levenskwaliteit
In onderzoek UC-II resulteerde behandeling met adalimumab in een verbetering van de score voor de ziektespecifieke inflammatoire darmziekte (IBDQ)-score.
immunogeniciteit
De vorming van anti-adalimumab-antilichamen is geassocieerd met een verhoogde klaring en verminderde werkzaamheid van adalimumab. Er is geen duidelijke correlatie tussen de aanwezigheid van anti-adalimumab-antilichamen en het optreden van bijwerkingen.
Patiënten in de onderzoeken naar reumatoïde artritis werden met verschillende tijdsintervallen gescreend op antilichamen tegen adalimumab gedurende de periode van 6 tot 12. In de klinische hoofdonderzoeken werden antilichamen tegen adalimumab gedetecteerd bij 5,5% (58/1053) van de patiënten die met adalimumab werden behandeld met 0,5% (2/370) van de patiënten behandeld met placebo Bij patiënten die niet gelijktijdig methotrexaat kregen, was de incidentie 12,4%, vergeleken met 0,6% wanneer adalimumab werd gebruikt in combinatie met methotrexaat.
Bij patiënten met polyarticulaire juveniele idiopathische artritis in de leeftijd van 4-17 jaar werden anti-adalimumab-antilichamen geïdentificeerd bij 15,8% van de patiënten (27/171) die werden behandeld met adalimumab. Bij patiënten die geen methotrexaat met Humira kregen, was de incidentie 25,6% (22/86) versus 5,9% (5/85) wanneer adalimumab werd gebruikt in combinatie met methotrexaat.
Bij patiënten met enthesitis-geassocieerde artritis werden anti-adalimumab-antilichamen geïdentificeerd bij 10,9% (5/46) van de met adalimumab behandelde patiënten. Bij patiënten die methotrexaat niet gelijktijdig met Humira kregen, was de incidentie 13,6% (3/22), vergeleken met 8,3% (2/24) wanneer adalimumab werd gebruikt in combinatie met methotrexaat.
Bij patiënten met artritis psoriatica werden anti-adalimumab-antilichamen geïdentificeerd bij 38/376 proefpersonen (10%) die met adalimumab werden behandeld. Bij patiënten die niet gelijktijdig met methotrexaat werden behandeld, was de incidentie 13,5% (24/178 proefpersonen) vergeleken met 7% (14 van de 198 proefpersonen) wanneer adalimumab werd gebruikt in combinatie met methotrexaat.
Bij patiënten met spondylitis ankylopoetica werden anti-adalimumab-antilichamen geïdentificeerd bij 17/204 proefpersonen (8,3%) die met adalimumab werden behandeld. Bij patiënten die niet gelijktijdig met methotrexaat werden behandeld, was de incidentie 16/185 (8,6%) vergeleken met 1/19 (5,3%) wanneer adalimumab werd gebruikt in combinatie met methotrexaat.
Bij patiënten met de ziekte van Crohn werden anti-adalimumab-antilichamen geïdentificeerd bij 7/269 proefpersonen (2,6%) en bij 19/487 proefpersonen bij patiënten met colitis ulcerosa.
Bij psoriasispatiënten werden anti-adalimumab-antilichamen geïdentificeerd bij 77/920 (8,4%) proefpersonen die werden behandeld met alleen adalimumab.
Bij patiënten met plaque psoriasis die langdurig als monotherapie met adalimumab werden behandeld en die deelnamen aan een onderzoek naar stopzetting en herbehandeling, was het percentage anti-adalimumab-antilichaampositief na hervatting van de behandeling (11 gevallen van de 482 proefpersonen, 2,3%) vergelijkbaar met dat waargenomen vóór stopzetting van het geneesmiddel (11 gevallen van de 590 proefpersonen, 1,9%).
Aangezien immunogeniciteitstesten productspecifiek zijn, is vergelijking van antilichaamhoeveelheden met andere producten niet geschikt.
Pediatrische populatie
Het Europees Geneesmiddelenbureau heeft besloten af te zien van de verplichting om de resultaten in te dienen van onderzoek dat is uitgevoerd met Humira bij alle subgroepen van pediatrische patiënten met reumatoïde artritis, artritis psoriatica en spondylitis ankylopoetica, zie rubriek 4.2 voor informatie over pediatrisch gebruik.
Het Europees Geneesmiddelenbureau heeft besloten tot uitstel van de verplichting voor de fabrikant om de resultaten in te dienen van onderzoek dat is uitgevoerd met Humira bij een of meer subgroepen van pediatrische patiënten met colitis ulcerosa, zie rubriek 4.2 voor informatie over pediatrisch gebruik.
05.2 Farmacokinetische eigenschappen
Absorptie en distributie
Na subcutane toediening van een enkelvoudige dosis van 40 mg was de absorptie en distributie van adalimumab traag, met piekserumconcentraties die ongeveer 5 dagen na toediening optraden. De gemiddelde absolute biologische beschikbaarheid van adalimumab in de drie onderzoeken na één subcutane dosis van 40 mg was 64 % Na enkelvoudige intraveneuze doses van 0,25 tot 10 mg/kg waren de concentraties dosisproportioneel Na doses van 0,5 mg/kg (≈40 mg) varieerde de klaring van 11 tot 15 ml/uur, varieerde het distributievolume (Vss) van 5 tot 6 liter, en de gemiddelde halfwaardetijd van de laatste fase was ongeveer twee weken. De concentraties van adalimumab in gewrichtsvloeistof bij verschillende patiënten met reumatoïde artritis varieerden van 31-96% van die in serum.
Na subcutane toediening van 40 mg adalimumab om de twee weken bij volwassen patiënten met reumatoïde artritis (RA), waren de dalconcentraties gemiddeld ongeveer 5 mcg/ml (zonder gelijktijdige behandeling met methotrexaat) en 8-9 mcg/ml (in combinatie met methotrexaat). serumspiegels van adalimumab bij evenwicht na subcutane doses van 20, 40 en 80 mg elke 2 weken of wekelijks stegen op een bijna dosisafhankelijke manier.
Na subcutane toediening van adalimumab 24 mg/m2 (tot een maximum van 40 mg) om de twee weken aan patiënten van 4 tot 17 jaar met polyarticulaire juveniele idiopathische artritis (JIA), is de gemiddelde minimale serumconcentratie baseline adalimumab (waarden gemeten vanaf week 20 tot en met week 48) was 5,6 ± 5,6 mcg/ml (102% CV) met adalimumab zonder gelijktijdig gebruik van methotrexaat en 10,9 ± 5,2 mcg/ml (47,7% CV) met gelijktijdig toegediende methotrexaat.
Bij patiënten met polyarticulaire JIA in de leeftijd van 2 tot 2 was de gemiddelde dal-evenwichtserumconcentratie van adalimumab 6,0 ± 6,1 g/ml (101% CV) met adalimumab zonder gelijktijdig gebruik van methotrexaat en 7,9 ± 5,6 mcg/ml (71,2% CV). bij gelijktijdige toediening met methotrexaat.
Na subcutane toediening van 24 mg/m2 (tot een maximum van 40 mg) om de twee weken aan patiënten van 6 tot 17 jaar met enthesitis-geassocieerde artritis, zijn de gemiddelde minimale serumconcentraties van adalimumab in het stadium van steady-state (gemeten waarden in week 24) waren 8,8 ± 6,6 mcg/ml met adalimumab zonder gelijktijdig gebruik van methotrexaat en 11,8 ± 4,3 mcg/ml bij gelijktijdige toediening met methotrexaat.
Bij psoriasispatiënten bleek de dalconcentratie gemiddeld ongeveer 5 mcg/ml te zijn tijdens behandeling met een dosis van 40 mg adalimumab eenmaal per twee weken toegediend als monotherapie.
Bij patiënten met de ziekte van Crohn bereikte de oplaaddosis met Humira 80 mg in week 0 gevolgd door Humira 40 mg in week 2 dalconcentraties van adalimumab in serum van ongeveer 5,5 mcg/ml tijdens de inductieperiode. Een oplaaddosis Humira 160 mg in week 0 gevolgd door Humira 80 mg in week 2 bereikte tijdens de inductieperiode dalconcentraties van adalimumab in het serum van ongeveer 12 mcg/ml. Gemiddelde evenwichtsniveaus van ongeveer 7 mcg/ml werden waargenomen bij patiënten met de ziekte van Crohn die elke twee weken een onderhoudsdosis van 40 mg Humira kregen.
Bij pediatrische patiënten met matige tot ernstige CD was de inductiedosis open-label adalimumab respectievelijk 160/80 mg of 80/40 mg in week 0 en 2, afhankelijk van de grenswaarde voor lichaamsgewicht bij 40 kg. In week 4 werden patiënten 1: 1 gerandomiseerd op basis van lichaamsgewicht in de behandelingsgroep met standaarddosering (40/20 mg eenmaal per twee weken) of lage dosering (20/10 mg eenmaal per twee weken). Gemiddelde (± SD) serumconcentraties van adalimumab-dalspiegels bereikt in week 4 waren 15,7 ± 6,6 mcg/ml voor patiënten ≥ 40 kg (160/80 mg) en 10,6 ± 6,1 mcg/ml voor proefpersonen
Voor patiënten die hun gerandomiseerde therapie bleven volgen, waren de gemiddelde (± SD) dalconcentraties van adalimumab in week 52 9,5 ± 5,6 mcg/ml voor de standaarddosisgroep en 3,5 ± 2,2 mcg/ml voor de lage dosisgroep. De gemiddelde dalconcentraties werden gehandhaafd bij patiënten die gedurende 52 weken om de twee weken werden behandeld met adalimumab. Voor patiënten die de dosis verhoogden van een regime om de twee weken naar een week ≤ waren de gemiddelde (± SD) serumconcentraties van adalimumab in week 52 15,3 ± 11,4 g/ml (40/20 mg, per week) en 6,7 ± 3,5 mcg/ ml (20/10 mg, per week).
Bij patiënten met colitis ulcerosa bereikte een oplaaddosis van 160 mg Humira in week 0 gevolgd door 80 mg Humira in week 2 een dalconcentratie van adalimumab van ongeveer 12 mcg/ml tijdens de inductieperiode. Gemiddelde evenwichtsniveaus van ongeveer 8 mcg/ml werden waargenomen bij patiënten met colitis ulcerosa die om de twee weken een onderhoudsdosis Humira 40 mg kregen.
Eliminatie
Farmacokinetische populatieanalyses op een steekproef van meer dan 1.300 RA-patiënten toonden een trend in de richting van een duidelijke toename van de adalimumabklaring naarmate het lichaamsgewicht toeneemt. Na correctie voor lichaamsgewicht bleken geslachts- en leeftijdsverschillen een minimaal effect te hebben op de adalimumabklaring. vrij adalimumab (niet gebonden aan anti-adalimumab antilichamen - AAA) waren lager bij patiënten met meetbare AAA-titers Humira is niet onderzocht bij patiënten met nierinsufficiëntie of leverinsufficiëntie.
Lever- of nierinsufficiëntie
Humira is niet onderzocht bij patiënten met nier- of leverinsufficiëntie.
05.3 Gegevens uit het preklinisch veiligheidsonderzoek
Niet-klinische gegevens duiden niet op een speciaal risico voor mensen. Deze gegevens zijn afkomstig van onderzoeken naar toxiciteit bij enkelvoudige doses, toxiciteit bij meervoudige doses en genotoxiciteit.
Een embryo-foetale ontwikkeling / perinatale ontwikkelingstoxiciteitsstudie werd uitgevoerd bij cynomologische apen met doses van 0, 30 en 100 mg / kg (9-17 apen / groep); deze studie bracht geen schade aan de foetus aan het licht die werd veroorzaakt door adalimumab. Carcinogeniteitstesten en standaard beoordelingen van vruchtbaarheid en postnatale toxiciteit werden niet uitgevoerd vanwege het ontbreken van geschikte modellen voor een antilichaam met beperkte kruisreactiviteit op TNF bij knaagdieren en de ontwikkeling van neutraliserende antilichamen bij knaagdieren.
06.0 FARMACEUTISCHE INFORMATIE
06.1 Hulpstoffen
Mannitol
Citroenzuur monohydraat
Natriumcitraat
Natriummonobasisch fosfaatdihydraat
Dinatriumfosfaatdihydraat
Natriumchloride
Polysorbaat 80
Natriumhydroxide
Water voor injecties.
06.2 Incompatibiliteit
Bij gebrek aan onderzoek naar onverenigbaarheden, mag dit geneesmiddel niet worden gemengd met andere geneesmiddelen.
06.3 Geldigheidsduur
24 maanden
06.4 Speciale voorzorgsmaatregelen bij bewaren
Bewaren in de koelkast (2 ° C - 8 ° C). Niet bevriezen. Bewaar de voorgevulde pen in de doos om het geneesmiddel tegen licht te beschermen.
Een enkele Humira-pen kan worden bewaard bij temperaturen tot maximaal 25°C voor een periode van maximaal 14 dagen. De pen moet worden beschermd tegen licht en weggegooid worden als hij gedurende de periode van 14 dagen niet wordt gebruikt.
06.5 Aard van de primaire verpakking en inhoud van de verpakking
Humira 40 mg oplossing voor injectie zit in voorgevulde wegwerppennen, klaar voor gebruik door de patiënt, met een voorgevulde spuit De spuit in de pen is van type I glas met een zuiger (broombutylrubber) en een naald met een dop (thermoplastische elastomeer).
verpakkingen:
• 1 voorgevulde pen met 1 alcoholdoekje in een blisterverpakking.
• 2 voorgevulde pennen, elk met 1 alcoholdoekje, in een blisterverpakking.
• 4 voorgevulde pennen, elk met 1 alcoholdoekje, in een blisterverpakking.
• 6 voorgevulde pennen, elk met 1 alcoholdoekje, in een blisterverpakking.
Mogelijk worden niet alle verpakkingsgrootten in de handel gebracht.
06.6 Instructies voor gebruik en verwerking
Humira 40 mg oplossing voor injectie bevat geen conserveermiddelen. Ongebruikte geneesmiddelen en afvalproducten van dit geneesmiddel moeten worden weggegooid in overeenstemming met de lokale regelgeving.
07.0 HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
AbbVie Ltd
maagdenkop
SL6 4XE
VK
08.0 NUMMER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
EU / 1/03/256/0071 voorgevulde pen 0,8 ml + 1 buffer
035946072 / E
EU / 1/03/256/0082 voorgevulde pennen 0,8 ml + 2 pads
035946084 / E
EU / 1/03/256/0094 voorgevulde pennen 0,8 ml + 4 pads
035946096 / E
EU / 1/03/256/0106 voorgevulde pennen 0,8 ml + 6 wattenstaafjes
035946108 / E
09.0 DATUM VAN EERSTE VERGUNNING OF VERLENGING VAN DE VERGUNNING
Datum eerste vergunning: 08 september 2003
Datum van laatste verlenging: 08 september 2008
10.0 DATUM VAN HERZIENING VAN DE TEKST
09/2014