Algemeenheid
Het syndroom van Crouzon is een zeldzame genetische ziekte, die de aanwezigheid van craniosynostose en andere nogal eigenaardige gezichtsafwijkingen bepaalt.
Het uiterlijk wordt veroorzaakt door bepaalde veranderingen in het DNA dat de FGFR2- en FGFR3-genen vormt; deze genetische elementen zijn betrokken bij het botrijpingsproces tijdens de embryonale ontwikkeling.

De therapie bestaat uit een reeks chirurgische ingrepen, gericht op het oplossen van de belangrijkste en gevaarlijkste symptomen.
Momenteel is de prognose vaak zeer positief.
Beoordeling van genetica
Voordat we verder gaan met de beschrijving van het Crouzon-syndroom, is het nuttig om enkele fundamentele concepten van genetica te bekijken.
Wat is DNA? Het is het genetische erfgoed, waarin de somatische eigenschappen, de aanleg, de fysieke kwaliteiten, het karakter, enz. van een levend organisme zijn geschreven. Het is vervat in alle cellen van het lichaam met een kern, zoals ligt hierin.
Wat zijn chromosomen? Volgens de definitie zijn chromosomen de structurele eenheden waarin DNA is georganiseerd. Menselijke cellen bevatten in hun kern 23 paren homologe chromosomen (22 van het autosomale niet-seksuele type en één paar van het seksuele type); elk paar is anders dan het andere, omdat het een specifieke gensequentie bevat.
Wat zijn genen? Het zijn korte stukjes DNA, of sequenties, van DNA met een fundamentele biologische betekenis: daaruit worden in feite eiwitten of biologische moleculen afgeleid die fundamenteel zijn voor het leven. In de genen zit een "geschreven" deel van wie we zijn en wie we zullen worden.
Elk gen is aanwezig in twee versies, de allelen: één allel is van moederlijke oorsprong, dus overgedragen door de moeder; het andere allel is van vaderlijke oorsprong en wordt daarom door de vader overgedragen.
Wat is een genetische mutatie Het is een fout in de DNA-sequentie, die een gen vormt. Door deze fout is het resulterende eiwit defect of volledig afwezig.In beide gevallen kunnen de effecten schadelijk zijn voor zowel het leven van de cel waarin de mutatie optreedt als dat van het organisme als geheel. Aangeboren ziekten en neoplasmata (dwz tumoren) behoren tot een of meer genetische mutaties.
Dat geldt ook voor het syndroom van Crouzon
Crouzon-syndroom is een zeldzame genetische aandoening die wordt gekenmerkt door craniosynostose en de "onnatuurlijke ontwikkeling van bepaalde gezichtselementen, waaronder de ogen, neus, kaak en kaak."
Het is een aangeboren ziekte waarvan de typische kenmerken al tijdens de eerste levensmomenten zichtbaar zijn.
BETEKENIS VAN CRANIOSINOSTOSI
Craniosynostose is de term waarmee artsen verwijzen naar de voortijdige fusie van een of meer schedelhechtingen.

Van de site: thecraniofacialcenter.com
Craniale hechtingen zijn de fibreuze gewrichten die de botten van het schedelgewelf met elkaar verbinden (d.w.z. de frontale, temporale, pariëtale en occipitale botten).
Onder normale omstandigheden vindt de fusie van de schedelhechtingen plaats in de postnatale periode (sommige processen eindigen zelfs op de leeftijd van 20). Door dit lange proces van fusie kunnen de hersenen voldoende groeien en zich ontwikkelen.
Als, zoals in het geval van craniosynostosen, de fusie te vroeg plaatsvindt - dus tijdens prenatale, perinatale * of vroege kinderjaren - ondergaan de hersenelementen (hersenen, cerebellum en hersenstam) en sommige zintuigen (met name ogen) een "verandering van vorm en groei.
* De perinatale term verwijst naar de levensfase die loopt van de 27e zwangerschapsweek tot de eerste 28 dagen na de bevalling.
OORSPRONG VAN DE NAAM
Het Crouzon-syndroom dankt zijn naam aan de Franse arts Octave Crouzon, die de verdienste heeft om eerst de belangrijkste klinische kenmerken ervan te hebben beschreven.
Crouzon leefde tussen de late jaren 1800 en vroege jaren 1900, precies van 1874 tot 1938. Aanvankelijk gebruikte hij de term craniofaciale dysostose om het syndroom te definiëren dat later zijn naam kreeg.
Oorzaken
Crouzon-syndroom ontstaat als gevolg van een mutatie in het FGFR2-gen, gelokaliseerd op chromosoom 10, of het FGFR3-gen, gelokaliseerd op chromosoom 4.
FGFR is het Engelse acroniem voor Fibroblast Groeifactor Receptor, wat in het Italiaans vertaald is: Receptor voor de fibroblastgroeifactor.
De functionele rol van de FGFR2- en FGFR3-genen is elk om een receptoreiwit te produceren, dat op zijn beurt de taak heeft om de rijping en embryonale ontwikkeling van botweefsel te reguleren.
Volgens de theorieën van de onderzoekers zouden de mutaties in FGFR2 en FGFR3 dezelfde genen hyperstimuleren, die, opnieuw actief, een vroege rijping van sommige botweefsels zouden induceren, inclusief die waaruit de schedel bestaat.
GENETICA
De genetische mutaties die verantwoordelijk zijn voor het syndroom van Crouzon kunnen erfelijk zijn of spontaan ontstaan na de conceptie.
In het eerste geval heeft de morbide aandoening - die artsen ook wel het erfelijke syndroom van Crouzon noemen - alle kenmerken van een autosomaal dominante genetische ziekte (of erfelijke dominante ziekte). Voor een beginnende lezer van genetica betekent dit dat:
- De ziekte en zijn symptomen komen ook voor in de aanwezigheid van slechts één gemuteerd gen-allel (het maakt niet uit of het van de moeder of de vader komt), aangezien de laatste dominant is over de gezonde.
- Een ouder die de mutatie draagt, is voldoende om de ziekte in een deel van het nageslacht te krijgen.
- De kans dat er een ziek kind wordt geboren uit een koppel waarbij slechts één van de twee componenten de mutatie draagt, is 50%.
In het tweede geval is de morbide aandoening - die experts aangeven met de terminologie van het niet-erfelijke syndroom van Crouzon - het resultaat van een abnormale sporadische gebeurtenis, die het DNA verandert tijdens de embryonale groei van de foetus.
Samenvatting van de betekenis van de termen erfelijk, autosomaal en dominant
- Erfelijk: het betekent dat de ouders de genetische wijziging die verantwoordelijk is voor de ziekte doorgeven aan het nageslacht (dwz aan de kinderen).
- Autosomaal: het betekent dat de mutatie die verantwoordelijk is voor de ziekte zich in een niet-geslachtschromosoom bevindt, dus autosomaal.
- Dominant: betekent dat de ziekte symptomen en tekenen veroorzaakt, zelfs wanneer slechts één allel van het verantwoordelijke gen is gemuteerd. In eenvoudiger bewoordingen is het alsof het allel met de mutatie meer kracht heeft dan het gezonde allel.
EPIDEMIOLOGIE
Volgens sommige schattingen van de incidentie van het Crouzon-syndroom zou ongeveer één op de 60.000 kinderen met deze zeldzame aandoening worden geboren.
Het Crouzon-syndroom is verantwoordelijk voor 4,5% van de gevallen van craniosynostose.
Symptomen en complicaties
Patiënten met het Crouzon-syndroom hebben een zeer specifiek symptoombeeld, dat meestal bestaat uit:
- Problemen met betrekking tot craniosynostose, waaronder:
-

Van https://en.wikipedia.org/wiki/Plagiocephaly brachycefalie, dat is het knijpen van de achterkant van het hoofd. Voortijdige fusie van de coronale schedelhechtingen volgt (coronale craniosynostose).
Indien onbehandeld, kan het de hersengroei en de ontwikkeling van cognitieve vaardigheden beïnvloeden.
Ze vertegenwoordigen een "alternatief voor brachycefalie: trigonocephalie (fusie van de metopische hechtdraad), dolichocephalie (fusie van de sagittale hechtdraad) en plagiocefalie (fusie van de coronale hechtingen). - Exophthalmus, wat de term is voor het uitpuilen van de oogbollen. Het kan de aanwezigheid van problemen met het gezichtsvermogen impliceren.
- Oculair hypertelorisme, dat wil zeggen, ogen die overdreven ver van elkaar verwijderd zijn. Met exophthalmus kan het zichtproblemen verergeren.
- misvormde neus, meestal in de vorm van een snavel. Indien ernstig of niet operatief behandeld, kan deze afwijking leiden tot ademhalingsproblemen of dezelfde symptomen als het obstructieve slaapapneusyndroom.
- Verhoogde intracraniale druk. Het is ook bekend als intracraniële hypertensie. Zijn aanwezigheid wordt verklaard door het feit dat de hersenstructuren niet de juiste ruimte hebben om te groeien.
Meestal gevonden in het midden van de late kinderjaren, is intracraniële hypertensie een mogelijke oorzaak van hoofdpijn, braken en oogpijn. - Hydrocephalus, wat het gevolg is van een toename van de cerebrospinale vloeistof in de subarachnoïdale ruimte en in de hersenventrikels.
- Arnold-Chiari-misvorming (of Arnold-Chiari-syndroom). Het is een afwijking aan de basis van de schedel.
* Hydrocephalus en Arnold-Chiari-misvorming zijn over het algemeen twee complicaties die optreden bij afwezigheid van adequate behandelingen. - Afwijkingen in de onderkaak en bovenkaak.
De eerste heeft kleinere afmetingen dan normaal, terwijl de tweede de neiging heeft om naar buiten uit te steken.Dit alles verandert de vorm van het gehemelte en de gebitsstructuur (afwezigheid van enkele tanden, enz.), met (soms zelfs ernstige) gevolgen voor de fonatie en op de kauwen.
Sommige patiënten worden geboren met een gespleten lip (gespleten lip) of een gespleten gehemelte.
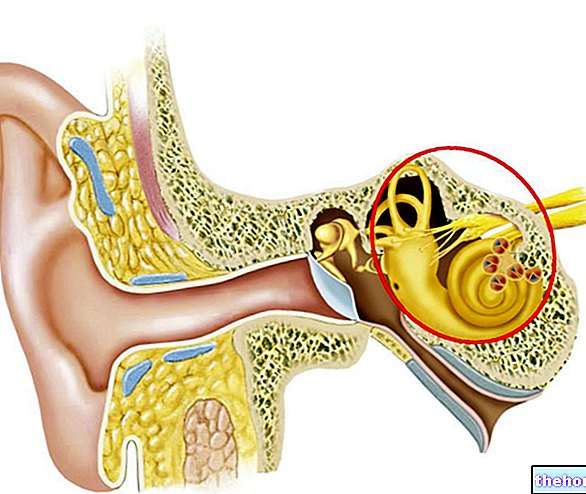
- Gehoorproblemen.
55% van de patiënten met het syndroom van Crouzon wordt geboren zonder gehoorgang of met grote afwijkingen daarin. Dit resulteert in een afwezige of sterk verminderde akoestische capaciteit.
Sommige proefpersonen ontwikkelen een reeks gehoorproblemen op volwassen leeftijd, toe te schrijven aan het typische klinische beeld van het syndroom van Ménière.
- Gewrichtsproblemen in de nek.
Ze betreffen 30% van de gevallen van het Crouzon-syndroom.
- Huidafwijkingen.
Patiënten met gemuteerd FGFR3-ondersteund Crouzon-syndroom aanwezig acanthosis nigricans, een dermatose die wordt gekenmerkt door een toename van de dikte (hyperkeratose) en donker worden (hyperpigmentatie) van de huid.

Twee andere anatomische anomalieën die (zij het zelden) geassocieerd zijn met het Crouzon-syndroom
- Patent arteriële duct
- Coarctatie van de aorta
CROUZON-SYNDROOM EN IQ
Mede dankzij de huidige mogelijkheden om craniosynostose te behandelen, heeft vandaag 97% van de patiënten met het Crouzon-syndroom een "normale intelligentie".
Diagnose
Een ervaren kinderarts kan het syndroom van Crouzon misschien alleen diagnosticeren door middel van lichamelijk onderzoek.
In het geval van enige twijfel of verbijstering zijn de volgende factoren van fundamenteel belang om tot een nauwkeurige conclusie te komen:
- Radiologische beelden, geleverd door röntgenfoto's of CT-scans van het hoofd
- Een genetische test, gericht op het zoeken naar eventuele DNA-mutaties.
OBJECTIEF ONDERZOEK
Het lichamelijk onderzoek bestaat uit een nauwkeurige analyse van het hoofd en de aanwezige afwijkingen.
Craniale misvormingen, veroorzaakt door craniosynostose (bijvoorbeeld brachycefalie), behoren tot de meest karakteristieke klinische symptomen van het syndroom van Crouzon en waarop de arts een deel van zijn diagnostische conclusies baseert.
RADIOLOGISCHE ONDERZOEKEN
Röntgenfoto's en CT-scans van het hoofd laten zien welke schedelhechtingen voortijdig zijn samengesmolten.
De craniosynostose die het syndroom van Crouzon kenmerkt, treft de coronale hechtingen, daarom is een gevonden fusie op het niveau van de laatste een vaak beslissende informatie voor diagnostische doeleinden.
GENETISCH ONDERZOEK
Naast het aantonen of het DNA mutaties heeft, helpt genetische testen bij het identificeren van het exacte gen dat het Crouzon-syndroom veroorzaakt, of dit nu FGFR2 of FGFR3 is.
Behandeling
Tegenwoordig kunnen dragers van het syndroom van Crouzon rekenen op verschillende behandelingen, afhankelijk van de ernst van de aandoening en de symptomen.
In feite hebben de artsen ervoor gezorgd:
- Chirurgie voor het oplossen van craniosynostose en de symptomen ervan.
- Akoestische hulpmiddelen, bij gehoorproblemen.
- Therapieën ter verbetering van de taalvaardigheid.
- Chirurgische therapieën voor de verbetering van anomalieën in de bovenkaak en onderkaak.
- Een operatie, ook wel een tracheostomie genoemd, om ademhalingsproblemen op te lossen.
Houd er rekening mee dat: Het syndroom van Crouzon is een morbide aandoening die voortkomt uit een "genetische verandering van het DNA die onmogelijk te genezen is. Dus in feite behandelen artsen de ziekte alleen vanuit een symptomatologisch oogpunt.
CHIRURGIE VOOR CRANIOSYNOSTOSE
De therapeutische doelstellingen van de chirurgische ingreep zijn twee:
- Geef de hersenstructuren en ogen de ruimte die ze nodig hebben om zich optimaal te ontwikkelen en te functioneren.
- Geef het hoofd een normale vorm en los vervolgens het probleem van brachycefalie op.
Chirurgen hebben de mogelijkheid om de operatie op twee verschillende manieren (of benaderingen) uit te voeren: via een "traditionele chirurgische ingreep - ook wel "open" genoemd - of via een" endoscopische chirurgische ingreep.
De "open operatie" omvat de "uitvoering van een" incisie op het hoofd, waardoor de opererende arts het bot of de misvormde schedelbeenderen extraheert die moeten worden geremodelleerd. Aan het einde van de remodellering plaatst de chirurg de eerder verwijderde botstructuren terug en sluit de incisie met hechtingen.
De endoscopische chirurgie daarentegen omvat het gebruik van een endoscoop en het oefenen van een zeer kleine incisie op het hoofd, waardoor de operatieve arts de endoscoop zelf inbrengt.
De endoscoop is in feite een dunne en flexibele buis, voorzien van een glasvezelcamera (aan het uiteinde in de schedel gestoken) en aangesloten op een monitor. Door dit specifieke instrument en de beelden die het op de monitor projecteert, is de chirurg in staat om de fuserende schedelhechtingen voortijdig te scheiden, met opmerkelijke precisie en zonder toevlucht te nemen tot huidincisies en botextracties.
Volgens deskundigen is de beste tijd om de operatie uit te voeren tijdens de zeer vroege kinderjaren (eerste 12 maanden van het leven), omdat de botten gemakkelijker worden gevormd.
Houd er echter rekening mee dat hoe jonger de patiënt is, hoe groter de kans op herhaling van dezelfde schedelnaden (recidief) In geval van recidief moet de operatie worden herhaald.
Volgens sommige statistische onderzoeken moet 10-20% van de zeer jonge proefpersonen die een craniosynostose-operatie ondergaan, een tweede operatie ondergaan vanwege een terugval.
BEHANDELING VAN AKOESTISCHE PROBLEMEN
Naast het voorschrijven van het gebruik van hoortoestellen, bevelen artsen ook periodieke gehoorcontroles aan, omdat dit de beste manier is om verergering van bestaande problemen te voorkomen.
CHIRURGISCHE THERAPIEN VOOR KAAK- EN KAAKANOMALIES
De behandeling van maxillaire en mandibulaire anomalieën omvat chirurgie voor het opnieuw uitlijnen van de bovenkaak en/of onderkaak, enkele tandheelkundige behandelingen voor de plaatsing van de tandbogen en de operatie voor het oplossen van de hazenlip en/of gespleten gehemelte.
TRACHEOSTOMIE
De tracheostomie is de chirurgische ingreep waardoor de arts ter hoogte van de nek (waar de luchtpijp passeert) een doorgang creëert voor de lucht die bestemd is voor de longen. Hierdoor kunnen degenen die deze operatie ondergaan weer en correct ademen.
Om de lucht in de longen te brengen, hebt u een kleine buis nodig, een transcheostomiebuis genaamd, die de juiste maat heeft om in de luchtpijp te worden ingebracht.
Prognose
Over het algemeen hangt de prognose af van de ernst van de craniosynostose: als deze met goed resultaat te behandelen is, kunnen patiënten met het syndroom van Crouzon een bijna normaal leven leiden.











.jpg)

-o-circuit-training-(ct)---tipologie.jpg)












