Actieve ingrediënten: Eltrombopag
Revolade 12,5 mg filmomhulde tabletten
Revolade 25 mg filmomhulde tabletten
Revolade 50 mg filmomhulde tabletten
Revolade 75 mg filmomhulde tabletten
Waarom wordt Revolade gebruikt? Waar is het voor?
Revolade bevat eltrombopag, dat behoort tot een groep geneesmiddelen die trombopoëtinereceptoragonisten worden genoemd. Het wordt gebruikt om het aantal bloedplaatjes in het bloed te verhogen Bloedplaatjes zijn bloedcellen die bloedingen verminderen of voorkomen.
- Revolade wordt gebruikt voor de behandeling van een 'bloedstollingsstoornis' genaamd auto-immuun (idiopathische) trombocytopenische purpura (ITP) bij patiënten (ouder dan 1 jaar) die al andere geneesmiddelen (corticosteroïden of immunoglobulinen) hebben gebruikt die niet effectief waren.
ITP wordt veroorzaakt door een laag aantal bloedplaatjes in het bloed (trombocytopenie). Mensen met ITP hebben een hoger risico op bloedingen. Symptomen bij patiënten met ITP kunnen zijn: petechiën (kleine platte rode ronde vlekken op de huid), blauwe plekken, epistaxis (neusbloedingen), bloedend tandvlees en het niet onder controle kunnen krijgen van het bloeden bij snijwonden of wonden.
- Revolade kan ook worden gebruikt bij de behandeling van een laag aantal bloedplaatjes (trombocytopenie) bij patiënten met het hepatitis C-virus (HCV), als ze problemen hebben gehad met bijwerkingen tijdens het gebruik van interferon. Veel mensen met hepatitis C hebben een laag aantal bloedplaatjes. als gevolg van de ziekte zelf, maar ook vanwege sommige antivirale geneesmiddelen die worden gebruikt om de ziekte te behandelen.
- Revolade kan ook worden gebruikt voor de behandeling van volwassen patiënten met een laag aantal bloedcellen veroorzaakt door ernstige aplastische anemie (SAA).
Contra-indicaties Wanneer Revolade niet mag worden gebruikt
Neem Revolade niet in
- als u allergisch bent voor eltrombopag of voor één van de andere bestanddelen van dit geneesmiddel (welke stoffen bevat Revolade).
- Raadpleeg uw arts als u denkt dat dit op u van toepassing is.
Voorzorgen bij gebruik Wat u moet weten voordat u Revolade inneemt
Praat met uw arts voordat u Revolade inneemt:
- als u leverproblemen heeft. Mensen met een laag aantal bloedplaatjes en een chronische (langdurige) gevorderde leverziekte lopen meer risico op bijwerkingen, waaronder leverbeschadiging en levensbedreigende bloedstolsels. Als uw arts van mening is dat de voordelen van het gebruik van Revolade opwegen tegen de risico's, zult u tijdens de behandeling nauwlettend worden gecontroleerd.
- als u risico loopt op bloedstolsels in uw aderen of slagaders, of als u weet dat bloedstolsels in uw familie vaak voorkomen.
- U loopt mogelijk een verhoogd risico op vorming van bloedstolsels:
- als u van hoge leeftijd bent
- als u lang in bed moest blijven
- als u een tumor heeft
- als u de anticonceptiepil of hormoonvervangende therapie gebruikt
- als u onlangs een operatie heeft ondergaan of lichamelijk trauma heeft opgelopen - als u ernstig overgewicht heeft (obesitas)
- als je een roker bent
- als u een gevorderde chronische leverziekte heeft
- Als een van deze situaties op u van toepassing is, informeer dan uw arts voordat u met de behandeling begint. U mag Revolade niet gebruiken tenzij uw arts van mening is dat de verwachte voordelen opwegen tegen de risico's van stolselvorming.
- als u last heeft van staar (troebeling van de ooglens)
- als u een andere bloedaandoening heeft, zoals myelodysplastisch syndroom (MDS). Uw arts zal tests uitvoeren om te controleren of u deze bloedaandoening niet heeft voordat u Revolade gaat gebruiken. Als u MDS heeft en Revolade gebruikt, kan uw MDS verergeren.
- Vertel het uw arts als een van deze situaties op u van toepassing is.
Oogonderzoek
Uw arts zal u aanraden om te controleren op staar. Als u geen routine oogonderzoeken heeft, zal uw arts een regelmatig onderzoek plannen. Het kan ook worden gecontroleerd op bloedingen in of rond het netvlies (de laag lichtgevoelige cellen aan de achterkant van het oog).
Hij zal regelmatig examens nodig hebben
Voordat u begint met het innemen van Revolade, zal uw arts bloedonderzoek doen om uw bloedcellen, inclusief bloedplaatjes, te controleren. Deze tests zullen met tussenpozen worden herhaald terwijl u het geneesmiddel inneemt.
Bloedonderzoek voor leverfunctie
Revolade kan de resultaten van bloedonderzoeken die kunnen wijzen op leverbeschadiging veranderen - een verhoging van sommige leverenzymen, met name bilirubine en alanine/aspartaattransaminasen. Als u wordt behandeld met interferon in combinatie met Revolade voor de behandeling van een laag aantal bloedplaatjes als gevolg van hepatitis C, kunnen sommige leverproblemen erger worden.
U moet bloedonderzoek ondergaan om uw leverfunctie te controleren voordat u begint met het gebruik van Revolade en tijdens de behandeling. Het kan zijn dat u moet stoppen met het innemen van Revolade als de hoeveelheid van deze enzymen te veel toeneemt of als er fysieke tekenen van leverbeschadiging optreden.
- Lees de informatie "Leverproblemen" in rubriek 4 van deze bijsluiter
Bloedonderzoek voor het aantal bloedplaatjes
Als u stopt met het innemen van Revolade, zal uw aantal bloedplaatjes waarschijnlijk binnen een paar dagen weer dalen.Uw bloedplaatjesaantal zal worden gecontroleerd en uw arts zal u vertellen over passende voorzorgsmaatregelen.
Een zeer hoog aantal bloedplaatjes kan het risico op bloedstolsels verhogen, maar er kunnen zich ook bloedstolsels vormen bij een normaal of zelfs laag aantal bloedplaatjes. Uw arts zal de dosis Revolade aanpassen om ervoor te zorgen dat het aantal bloedplaatjes niet te veel stijgt.
Zoek onmiddellijk medische hulp als u een van deze tekenen van een bloedstolsel heeft:
- zwelling, pijn of gevoeligheid in één been
- plotseling begin van kortademigheid, vooral samen met scherpe pijn op de borst of snelle ademhaling
- abdominale (maag) pijn, zwelling van de buik, bloed in de ontlasting.
Tests om uw beenmerg te controleren
Bij mensen die mogelijk beenmergproblemen hebben, kunnen geneesmiddelen zoals Revolade de problemen verergeren. Tekenen van veranderingen in het beenmerg kunnen zich voordoen als afwijkingen in de resultaten van bloedonderzoek. Uw arts kan tests uitvoeren om uw beenmerg direct te controleren terwijl u met Revolade wordt behandeld.
Tests voor spijsverteringsbloedingen
Als u wordt behandeld met interferongeneesmiddelen in combinatie met Revolade, wordt u gecontroleerd op tekenen van bloeding in uw maag of darmen nadat u stopt met het innemen van Revolade.
Hart check
Uw arts kan het nodig vinden om uw hart te controleren tijdens de behandeling met Revolade en om een elektrocardiogram (ECG) uit te voeren.
Kinderen en adolescenten
Revolade wordt niet aanbevolen voor kinderen jonger dan 1 jaar met ITP. Het wordt ook niet aanbevolen bij mensen onder de 18 jaar met een laag aantal bloedplaatjes als gevolg van hepatitis C of ernstige aplastische anemie.
Interacties Welke medicijnen of voedingsmiddelen kunnen het effect van Revolade veranderen
Vertel uw arts of apotheker als u andere geneesmiddelen gebruikt, kort geleden heeft gebruikt of gaat gebruiken.
Sommige van de veelgebruikte geneesmiddelen hebben een wisselwerking met Revolade, waaronder geneesmiddelen op recept en zonder recept, en mineralen. Waaronder:? maagzuurremmers om indigestie, brandend maagzuur of maagzweren te behandelen Wanneer moet u het innemen)
- geneesmiddelen die statines worden genoemd, om cholesterol te verlagen
- sommige geneesmiddelen om een hiv-infectie te behandelen, zoals lopinavir en/of ritonavir
- ciclosporine gebruikt bij transplantaties en immuunziekten
- mineralen zoals ijzer, calcium, magnesium, aluminium, selenium en zink die te vinden zijn in vitamine- en mineralensupplementen (wanneer in te nemen)
- geneesmiddelen zoals methotrexaat en topotecan, om kanker te behandelen
- Vertel het uw arts als u een van deze geneesmiddelen gebruikt. Sommige kunnen niet samen met Revolade worden ingenomen, of de dosis die u inneemt, moet mogelijk worden aangepast of het tijdstip waarop u deze inneemt, moet worden gewijzigd. Uw arts zal alle geneesmiddelen die u inneemt beoordelen en indien nodig voorstellen dat u ze indien nodig vervangt.
Er is een hoger risico op bloedingen als u ook geneesmiddelen gebruikt om bloedstolsels te voorkomen. Uw arts zal dit met u bespreken.
Als u corticosteroïden, danazol en/of azathioprine gebruikt, moet u mogelijk een lagere dosis nemen of ermee stoppen terwijl u Revolade gebruikt.
Waarop moet u letten met eten en drinken
Neem Revolade niet in met dranken of zuivelproducten en kaas, aangezien het calcium in melkproducten de absorptie van het geneesmiddel beïnvloedt.
Waarschuwingen Het is belangrijk om te weten dat:
Zwangerschap en borstvoeding
Gebruik Revolade niet als u zwanger bent, tenzij uw arts dit specifiek aanbeveelt. Het effect van Revolade tijdens de zwangerschap is niet bekend.
- Vertel het uw arts als u zwanger bent, als u denkt dat u zwanger bent of als u van plan bent zwanger te worden.
- Gebruik een betrouwbare anticonceptiemethode terwijl u Revolade gebruikt om zwangerschap te voorkomen
- Als u zwanger wordt terwijl u Revolade gebruikt, vertel dit dan aan uw arts.
Geef geen borstvoeding terwijl u Revolade gebruikt. Het is niet bekend of Revolade in de moedermelk terechtkomt.
- Als u borstvoeding geeft of van plan bent borstvoeding te geven, vertel dit dan aan uw arts.
Rijvaardigheid en het gebruik van machines
- Revolade kan u duizelig maken en andere bijwerkingen hebben die leiden tot minder aandacht.
- Bestuur geen voertuigen en gebruik geen machines tenzij u zeker weet dat u er geen last van heeft.
Dosis, wijze en tijdstip van toediening Hoe Revolade te gebruiken: Dosering
Gebruik dit geneesmiddel altijd precies zoals uw arts u dat heeft verteld. Raadpleeg bij twijfel uw arts of apotheker. Verander uw dosis of uw schema van Revolade niet tenzij uw arts of apotheker u dit adviseert. Terwijl u Revolade gebruikt, wordt u behandeld door een arts met specialistische ervaring in de behandeling van uw aandoening.
Hoeveel te nemen?
Voor ITP
Volwassenen en kinderen (6 tot 17 jaar) - de aanbevolen startdosering voor ITP is één Revolade 50 mg tablet per dag.Als u van Oost-Aziatische afkomst bent (Chinees, Japans, Taiwanees, Thais of Koreaans), moet u mogelijk beginnen met een lagere dosis van 25 mg.
Kinderen (1 tot 5 jaar) - de aanbevolen startdosering voor ITP is één Revolade-tablet van 25 mg per dag.
Voor hepatitis C
Volwassenen - De aanbevolen startdosering voor hepatitis C is één Revolade-tablet van 25 mg per dag Als u van Oost-Aziatische afkomst bent (Chinees, Japans, Taiwanees, Thais of Koreaans), begint u met dezelfde dosis van 25 mg.
Voor SAA
Volwassenen - de aanbevolen startdosering voor AAS is één Revolade-tablet van 50 mg per dag. Als u van Oost-Aziatische afkomst bent (Chinees, Japans, Taiwanees, Thais of Koreaans), moet u mogelijk beginnen met een lagere dosis van 25 mg.
Revolade kan 1 tot 2 weken duren om te werken. Op basis van uw reactie op Revolade kan uw arts een dagelijkse dosisaanpassing aanbevelen.
Hoe de tabletten in te nemen?
Slik de tablet heel door met wat water.
Wanneer te nemen?
Zeker weten dat-
- in de 4 uur voor het innemen van Revolade
- en in de 2 uur na inname van Revolade
U verbruikt geen van de volgende producten:
- voedsel zoals kaas, boter, yoghurt of ijs
- melk of smoothies op basis van melk, dranken met melk, yoghurt of room
- antacida, een soort geneesmiddel tegen indigestie en brandend maagzuur
- sommige vitamine- en mineralensupplementen, waaronder ijzer, calcium, magnesium, aluminium, selenium en zink.
Als dit het geval is, wordt het geneesmiddel niet goed door het lichaam opgenomen.
Vraag uw arts voor meer advies over geschikt eten en drinken.
Bent u vergeten Revolade in te nemen?
Neem de volgende dosis op het gebruikelijke tijdstip. Neem niet meer dan één dosis Revolade per dag in.
Als u stopt met het gebruik van Revolade
Stop niet met het innemen van Revolade zonder met uw arts te overleggen. Als uw arts u adviseert de behandeling te stoppen, wordt uw aantal bloedplaatjes gedurende vier weken wekelijks gecontroleerd.
Als u nog vragen heeft over het gebruik van dit geneesmiddel, neem dan contact op met uw arts of apotheker.
Overdosering Wat moet u doen als u te veel Revolade heeft ingenomen?
Neem onmiddellijk contact op met uw arts of apotheker. Laat hen indien mogelijk de doos of deze bijsluiter zien. Eventuele tekenen of symptomen van bijwerkingen zullen worden gecontroleerd en onmiddellijk op de juiste manier worden behandeld.
Bijwerkingen Wat zijn de bijwerkingen van Revolade
Zoals alle geneesmiddelen kan ook dit geneesmiddel bijwerkingen hebben, al krijgt niet iedereen daarmee te maken.
Symptomen die aandacht nodig hebben: Raadpleeg een arts
Mensen die Revolade gebruiken voor ITP of een laag aantal bloedplaatjes als gevolg van hepatitis C, kunnen tekenen van mogelijk ernstige bijwerkingen krijgen. Het is belangrijk om het uw arts te vertellen als u deze symptomen krijgt.
Hoger risico op bloedstolsels
Sommige mensen hebben mogelijk een hoger risico op bloedstolsels en geneesmiddelen zoals Revolade kunnen dit probleem verergeren. Plotselinge verstopping van een bloedvat door een bloedstolsel is een bijwerking die soms voorkomt en kan optreden bij maximaal 1 op de 100 mensen.
Roep onmiddellijk medische hulp in als u tekenen en symptomen van een bloedstolsel ervaart, zoals:
- zwelling, pijn, warmte, roodheid of gevoeligheid in één been
- plotseling begin van kortademigheid, vooral samen met scherpe pijn op de borst of snelle ademhaling
- abdominale (maag) pijn, zwelling van de buik, bloed in de ontlasting.
Leverproblemen
Revolade kan veranderingen veroorzaken die blijken uit bloedonderzoeken en kunnen tekenen zijn van leverbeschadiging. Leverproblemen (verhogingen van enzymen gevonden in bloedtesten) komen vaak voor en kunnen voorkomen bij maximaal 1 op de 10 mensen Andere leverproblemen (gal die niet goed stroomt) komen soms voor en kunnen voorkomen bij maximaal 1 op de 10 mensen.
Als u een van deze tekenen van leverproblemen heeft:
- geel worden van de huid of het wit van de ogen (geelzucht)
- ongewoon donker gekleurde urine
- Vertel het uw arts onmiddellijk.
Bloeden of blauwe plekken na het stoppen van de behandeling
Binnen twee weken nadat u met Revolade bent gestopt, zal uw aantal bloedplaatjes gewoonlijk dalen tot wat het was voordat u met Revolade begon. Een lager aantal bloedplaatjes kan het risico op bloedingen of blauwe plekken vergroten. Uw arts zal uw aantal bloedplaatjes controleren gedurende ten minste 4 weken nadat u bent gestopt met het innemen van Revolade.
- Vertel het uw arts als u bloedingen of blauwe plekken krijgt nadat u met Revolade bent gestopt.
Sommige mensen hebben bloedingen in het spijsverteringsstelsel nadat ze zijn gestopt met het gebruik van peginterferon, ribavirine en Revolade. Symptomen zijn onder meer:
- donkere ontlasting, kleurverandering van de stoelgang is een soms voorkomende bijwerking die bij maximaal 1 op de 100 mensen kan optreden)
- bloed bij de ontlasting
- bloed overgeven of zoiets als koffiedik
- Vertel het uw arts onmiddellijk als u een van deze symptomen krijgt.
Andere mogelijke bijwerkingen bij volwassenen met ITP
Vaak voorkomende bijwerkingen Deze kunnen voorkomen bij maximaal 1 op de 10 mensen:
- misselijkheid
- diarree
- cataract (troebeling van de ooglens)
- droge ogen
- ongebruikelijk haarverlies of dunner worden
- uitslag
- jeuk
- spierpijn, spierspasmen
- rugpijn
- bot pijn
- tintelingen en gevoelloosheid in de handen of voeten
- zware menstruatiecyclus
- mondzweren.
Vaak voorkomende bijwerkingen die kunnen blijken uit een bloedonderzoek:
- verhoogde leverenzymen
- verhoogd bilirubine (een stof die door de lever wordt geproduceerd)
- verhoogde niveaus van sommige eiwitten.
Soms voorkomende bijwerkingen
Deze kunnen voorkomen bij maximaal 1 op de 100 mensen:
- onderbreking van de bloedtoevoer naar een deel van het hart
- plotselinge kortademigheid, vooral wanneer deze gepaard gaat met scherpe pijn op de borst en/of snelle ademhaling, wat een teken kan zijn van een bloedstolsel in de longen (zie "Hoger risico op bloedstolsels" aan het begin van rubriek 4
- functieverlies van een deel van de long veroorzaakt door verstopping van de longslagader
- leverproblemen, waaronder gele verkleuring van de ogen en huid
- snelle hartslag, onregelmatige hartslag, blauwachtige verkleuring van de huid
- hartritmestoornissen (QT-verlenging)
- ontsteking van een ader
- blauwe plekken
- keelpijn en ongemak bij het slikken, ontsteking van de longen, sinussen, amandelen, neus en keel
- invloed hebben
- longontsteking
- verlies van eetlust
- pijnlijke zwelling van de gewrichten veroorzaakt door urinezuur (jicht)
- slaapproblemen, depressie, verlies van interesse, stemmingswisselingen
- slaperigheid, evenwichtsproblemen, spraak- en zenuwfunctie, migraine, tremoren
- oogproblemen, waaronder wazig en minder helder zicht
- oorpijn, duizeligheid
- problemen met neus, keel en sinussen, ademhalingsproblemen tijdens de slaap
- problemen met het spijsverteringsstelsel, waaronder: braken, winderigheid, frequente stoelgang, buikpijn en gevoeligheid, voedselvergiftiging
- rectale kanker
- mondproblemen, waaronder een droge of pijnlijke mond, gevoeligheid van de tong, bloedend tandvlees,
- huidveranderingen waaronder overmatig zweten, blaren en jeukende uitslag, rode vlekken, veranderingen in uiterlijk
- zonnebrand
- roodheid of zwelling rond een wond
- bloeding rond een katheter (indien aanwezig)
- gevoel van een vreemd lichaam op de injectieplaats
- spier zwakte
- nierproblemen, waaronder: ontsteking van de nieren, 's nachts overmatig plassen, nierfalen, urineweginfectie, witte bloedcellen in de urine
- malaise, koorts, het warm hebben, pijn op de borst
- koud zweet
- ontsteking van het tandvlees
- huidinfectie.
Soms voorkomende bijwerkingen die kunnen blijken uit een bloedtest:
- afname van het aantal rode bloedcellen (bloedarmoede), witte bloedcellen en bloedplaatjes
- verhoogd aantal rode bloedcellen
- veranderingen in bloedmorfologie
- veranderingen in urinezuur-, calcium- en kaliumspiegels.
Andere mogelijke bijwerkingen bij kinderen met ITP
Zeer vaak voorkomende bijwerkingen
Deze kunnen voorkomen bij meer dan 1 op de 10 kinderen:
- keelpijn, loopneus, verstopte neus en niezen
- infectie van de neus, sinussen, keel en bovenste luchtwegen, verkoudheid (infectie van de bovenste luchtwegen)
- diarree.
Vaak voorkomende bijwerkingen
Deze kunnen voorkomen bij maximaal 1 op de 10 kinderen:
- moeite met slapen (slapeloosheid)
- buikpijn
- kiespijn
- hoest
- pijn in neus en keel
- jeukende neus, loopneus of verstopte neus
- hoge temperatuur.
Andere mogelijke bijwerkingen bij mensen met hepatitis C.
Zeer vaak voorkomende bijwerkingen
Deze kunnen voorkomen bij meer dan 1 op de 10 mensen:
- hoofdpijn
- verminderde eetlust
- slapeloosheid
- hoest
- misselijkheid, diarree
- spierpijn, jeuk, gebrek aan energie, hoge temperatuur, ongebruikelijk haarverlies, gevoel van zwakte, griepachtige ziekte, zwelling in handen en voeten, koude rillingen.
Zeer vaak voorkomende bijwerkingen die kunnen blijken uit een bloedtest:
- afname van het aantal rode bloedcellen (bloedarmoede).
Vaak voorkomende bijwerkingen
Deze kunnen voorkomen bij maximaal 1 op de 10 mensen:
- urineweginfecties
- ontsteking van de neusholtes, keel en mond, griepachtige symptomen, droge mond, pijnlijke of pijnlijke mond, kiespijn
- gewichtsverlies
- slaapstoornissen, abnormale slaperigheid, verwardheid, depressie, angst, opwinding
- duizeligheid, problemen met concentratie en geheugen
- tintelingen of gevoelloosheid in de handen of voeten
- ontsteking in de hersenen
- oogproblemen waaronder staar (troebeling van de ooglens) droge ogen, kleine gele afzettingen op het netvlies, geel worden van het oogwit
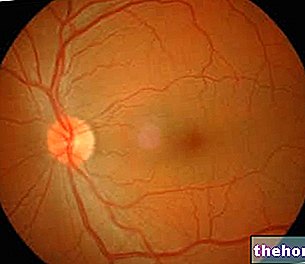
- bloeding in of rond het netvlies (aanwezig in de achterkant van het oog)
- gevoel van duizeligheid, hartkloppingen, kortademigheid
- hoesten met slijm
- problemen met het spijsverteringsstelsel, waaronder: braken, maagpijn, indigestie, constipatie, maagzwelling, smaakstoornis, maagontsteking, aambeien, gezwollen bloedvaten en bloeding in de slokdarm (oesofagitis), irritatie van de darm
- leverproblemen, waaronder: bloedstolsel, geel worden van het oogwit of de huid (geelzucht), leverkanker
- huidveranderingen, waaronder: uitslag, droge huid, eczeem, roodheid van de huid, jeuk, overmatig zweten, ongewone huidgroei? gewrichtspijn, rugpijn, botpijn, pijn in de handen of voeten, spierspasmen
- prikkelbaarheid, algemeen gevoel van onwel zijn, pijn op de borst en ongemak
- reacties op de injectieplaats
- hartritmestoornissen (QT-verlenging).
Vaak voorkomende bijwerkingen die kunnen blijken uit een bloedonderzoek:
- verhoogde bloedsuikerspiegel (hyperglykemie)
- afname van het aantal witte bloedcellen
- vermindering van bloedeiwitten
- afbraak van rode bloedcellen (hemolytische anemie)
- verhoogd bilirubine (een stof die door de lever wordt geproduceerd)
- veranderingen in de enzymen die de bloedstolling regelen.
Soms voorkomende bijwerkingen
Deze kunnen voorkomen bij maximaal 1 op de 100 mensen:
- pijn bij het plassen.
Soms voorkomende bijwerkingen
De frequentie kan met de beschikbare gegevens niet worden bepaald
- verkleuring van de huid
De volgende bijwerkingen zijn gemeld in verband met behandeling met Revolade bij patiënten met ernstige aplastische anemie (SAA).
Zeer vaak voorkomende bijwerkingen
Deze kunnen voorkomen bij maximaal 1 op de 10 mensen:
- hoest
- hoofdpijn
- piepende ademhaling (dyspneu)
- pijn in neus en keel
- een loop neus
- buikpijn
- diarree
- misselijkheid
- blauwe plekken
- gewrichtspijn
- spiertrekkingen
- pijn in de armen, benen, handen en voeten
- duizeligheid
- erg moe voelen
- koorts
- slapeloosheid
Zeer vaak voorkomende bijwerkingen die kunnen blijken uit een bloedtest:
- verhoogde leverenzymen (transaminasen). Bloedonderzoek kan abnormale veranderingen in de beenmergcellen laten zien.
Vaak voorkomende bijwerkingen
Deze kunnen voorkomen bij maximaal 1 op de 10 mensen:
- ongerustheid
- depressie
- Het koud hebben
- algemene malaise
- oogproblemen, waaronder: wazig en minder helder zien, cataracten, het zien van vlekken in het oog als gevolg van een onvolmaakte transparantie van het glasvocht, droge ogen, jeukende ogen, gele verkleuring van de huid of het wit van de ogen
- neusbloedingen
- bloedend tandvlees
- blaren in de mond
- problemen met het spijsverteringsstelsel, waaronder: braken, veranderingen in eetlust (toename of afname), maagpijn/ongemak, opgeblazen maag, winderigheid, verandering in kleur van de ontlasting
- flauwvallen
- huidproblemen waaronder: kleine rode of paarse vlekken veroorzaakt door bloedingen in de huid (petechiën), uitslag, jeuk, huidlaesies
- rugpijn
- pijn in de spieren
- bot pijn
- zwakheid
- zwelling van weefsels, meestal van de onderste ledematen, als gevolg van waterretentie
- abnormaal verkleurde urine
- verstoring van de bloedtoevoer naar de milt (miltinfarct).
Vaak voorkomende bijwerkingen die kunnen blijken uit een bloedonderzoek:
- verhoogde enzymen als gevolg van spierbeschadiging (creatinefosfokinase)
- ophoping van ijzer in het bloed
- verminderd aantal witte bloedcellen (neutropenie)
- verlaagde bloedsuikerspiegel (hypoglykemie)
- verhoogd bilirubine (een stof die door de lever wordt geproduceerd)
Soms voorkomende bijwerkingen
De frequentie kan met de beschikbare gegevens niet worden bepaald
- verkleuring van de huid
Melding van bijwerkingen
Krijgt u last van bijwerkingen, neem dan contact op met uw arts, apotheker of verpleegkundige. Dit geldt ook voor alle mogelijke bijwerkingen die niet in deze bijsluiter staan. U kunt bijwerkingen ook rechtstreeks melden via het nationale meldsysteem. Door bijwerkingen te melden, kunt u zorgen voor meer informatie over de veiligheid van dit geneesmiddel.
Vervaldatum en retentie
Buiten het zicht en bereik van kinderen houden.
Gebruik dit geneesmiddel niet meer na de uiterste houdbaarheidsdatum. Die is te vinden op de doos en blister na EXP. De uiterste houdbaarheidsdatum verwijst naar de laatste dag van die maand.
Voor dit geneesmiddel zijn er geen speciale bewaarcondities.
Gooi geneesmiddelen niet weg via het afvalwater of met huishoudelijk afval. Vraag uw apotheker wat u met geneesmiddelen moet doen die u niet meer gebruikt. Dit helpt het milieu te beschermen.
Andere informatie
Wat bevat Revolade?
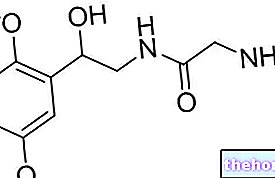
Het werkzame bestanddeel in Revolade is eltrombopag.
12,5 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 12,5 mg eltrombopag.
25 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 25 mg eltrombopag.
50 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 50 mg eltrombopag.
75 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 75 mg eltrombopag.
De andere stoffen in dit middel zijn: hypromellose, macrogol 400, magnesiumstearaat, mannitol (E421), microkristallijne cellulose, povidon, natriumzetmeelglycolaat, titaandioxide (E171).
Revolade 50 mg filmomhulde tabletten bevatten ook rood ijzeroxide (E172), geel ijzeroxide (E172).
Revolade 75 mg filmomhulde tabletten bevatten ook rood ijzeroxide (E172), geel ijzeroxide (E172).
Hoe ziet Revolade eruit en hoeveel zit er in een verpakking?
Revolade 12,5 mg filmomhulde tabletten zijn rond, biconvex, wit, met aan één kant de inscriptie "GS MZ1" en "12.5".
Revolade 25 mg filmomhulde tabletten zijn rond, biconvex, wit, met aan één kant de inscriptie "GS NX3" en "25".
Revolade 50 mg filmomhulde tabletten zijn rond, biconvex, bruin, met aan één kant "GS UFU" en "50".
Revolade 75 mg filmomhulde tabletten zijn rond, biconvex, roze, met aan één kant de inscriptie "GS FFS" en "75".
Ze worden geleverd in aluminium blisterverpakkingen in een verpakking met 14 of 28 filmomhulde tabletten en een multiverpakking met 84 (3 verpakkingen van 28) filmomhulde tabletten.
Mogelijk worden niet alle verpakkingsgrootten in uw land in de handel gebracht.
Bron Bijsluiter: AIFA (Italiaans Geneesmiddelenbureau). Inhoud gepubliceerd in januari 2016. De aanwezige informatie is mogelijk niet up-to-date.
Om toegang te hebben tot de meest actuele versie, is het raadzaam om de website van AIFA (Italian Medicines Agency) te bezoeken. Disclaimer en nuttige informatie.
01.0 NAAM VAN HET GENEESMIDDEL
REVOLADE TABLETTEN BEDEKT MET FILM
02.0 KWALITATIEVE EN KWANTITATIEVE SAMENSTELLING
Revolade 12,5 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 12,5 mg eltrombopag.
Revolade 25 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 25 mg eltrombopag.
Revolade 50 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 50 mg eltrombopag.
Revolade 75 mg filmomhulde tabletten
Elke filmomhulde tablet bevat eltrombopag olamine overeenkomend met 75 mg eltrombopag.
Voor de volledige lijst van hulpstoffen, zie rubriek 6.1.
03.0 FARMACEUTISCHE VORM
Filmomhulde tablet.
Revolade 12,5 mg filmomhulde tabletten
Ronde, biconvexe, witte filmomhulde tablet (ongeveer 7,9 mm in diameter) met aan één zijde de inscriptie "GS MZ1" en "12.5".
Revolade 25 mg filmomhulde tabletten
Ronde, biconvexe, witte filmomhulde tablet (ongeveer 10,3 mm in diameter) met aan één kant de inscriptie "GS NX3" en "25".
Revolade 50 mg filmomhulde tabletten
Ronde, biconvexe, bruine filmomhulde tablet (ongeveer 10,3 mm in diameter) met aan één kant de inscriptie "GS UFU" en "50".
Revolade 75 mg filmomhulde tabletten
Ronde, biconvexe, roze filmomhulde tablet (ongeveer 10,3 mm in diameter) met aan één kant de inscriptie "GS FFS" en "75".
04.0 KLINISCHE INFORMATIE
04.1 Therapeutische indicaties
Revolade is geïndiceerd bij patiënten ouder dan 1 jaar met chronische auto-immuun (idiopathische) trombocytopenische purpura (ITP) die ongevoelig zijn voor andere behandelingen (bijv. corticosteroïden, immunoglobulinen) (zie rubrieken 4.2 en 5.1).
Revolade is geïndiceerd bij volwassen patiënten met chronische hepatitis C-virusinfectie (Hepatitis C-virus, HCV) voor de behandeling van trombocytopenie, wanneer de mate van trombocytopenie de belangrijkste factor is die het starten verhindert of het vermogen om een optimale op interferon gebaseerde therapie te handhaven beperkt (zie rubrieken 4.4 en 5.1).
Revolade is geïndiceerd bij volwassen patiënten met ernstige verworven aplastische anemie (SAA), ongevoelig voor eerdere immunosuppressieve therapie of zwaar voorbehandeld en komen niet in aanmerking voor hematopoëtische stamceltransplantatie (zie rubriek 5.1).
04.2 Dosering en wijze van toediening
Behandeling met eltrombopag moet worden gestart en voortgezet onder toezicht van een arts die ervaring heeft met de behandeling van hematologische aandoeningen of met de behandeling van chronische hepatitis C en de complicaties daarvan.
Dosering
De vereiste dosering van eltrombopag moet individueel worden bepaald op basis van het aantal bloedplaatjes van de patiënt. Het doel van de behandeling met eltrombopag mag niet zijn om het aantal bloedplaatjes te normaliseren.
Het poeder voor orale suspensie kan leiden tot een hogere blootstelling aan eltrombopag dan de tabletformulering (zie rubriek 5.2). Bij het overschakelen tussen tablet- en poederformuleringen voor orale suspensie moet het aantal bloedplaatjes wekelijks gedurende 2 weken worden gecontroleerd.
Chronische auto-immuun (idiopathische) trombocytopenie
De laagste dosis eltrombopag moet worden gebruikt om een trombocytenaantal van ≥ 50.000 / l te bereiken en te behouden. Dosisaanpassingen zijn gebaseerd op de respons op het aantal bloedplaatjes.
Eltrombopag mag niet worden gebruikt om het aantal bloedplaatjes te normaliseren. In klinische onderzoeken nam het aantal bloedplaatjes over het algemeen toe binnen 1-2 weken na het starten met eltrombopag en daalde het binnen 1-2 weken na het stoppen.
Volwassenen en pediatrische patiënten van 6-17 jaar
De aanbevolen startdosering van eltrombopag is 50 mg eenmaal daags. Voor patiënten van Oost-Aziatische afkomst (zoals Chinees, Japans, Taiwanees, Koreaans of Thais), dient de behandeling met eltrombopag te worden gestart met een verlaagde dosis van 25 mg eenmaal daags (zie rubriek 5.2).
Pediatrische patiënten van 1 tot 5 jaar
De aanbevolen startdosering van eltrombopag is eenmaal daags 25 mg.
Dosisbewaking en -aanpassing
Na het starten van de behandeling met eltrombopag moet de dosis worden aangepast om een bloedplaatjesaantal van ≥ 50.000 / l te bereiken en te behouden dat nodig is om het risico op bloedingen te verminderen. De dagelijkse dosis van 75 mg mag niet worden overschreden.
Bloedchemie en leverfunctieparameters moeten regelmatig worden gecontroleerd tijdens de behandeling met eltrombopag en het doseringsschema van eltrombopag moet worden aangepast op basis van het aantal bloedplaatjes zoals vermeld in tabel 1. Tijdens de behandeling met eltrombopag moeten de volledige bloedtellingen wekelijks worden beoordeeld, inclusief het aantal bloedplaatjes en perifere bloeduitstrijkje, totdat een stabiel aantal bloedplaatjes is bereikt (≥ 50.000 / l gedurende minimaal 4 weken).
Vervolgens moet maandelijks een volledig bloedbeeld worden uitgevoerd, inclusief het aantal bloedplaatjes en een uitstrijkje van perifeer bloed.
Tabel 1 Dosisaanpassingen eltrombopag bij ITP-patiënten
* - Voor patiënten die eltrombopag 25 mg eenmaal per twee dagen gebruiken, de dosis verhogen tot 25 mg eenmaal per dag.
? - Voor patiënten die eltrombopag 25 mg eenmaal daags innemen, dient een dosis van 12,5 mg eenmaal daags of als alternatief een dosis van 25 mg eenmaal per twee dagen te worden overwogen.
Eltrombopag kan naast andere ITP-geneesmiddelen worden gegeven. Het doseringsschema van gelijktijdig toegediende geneesmiddelen voor de behandeling van ITP moet, indien klinisch aangewezen, worden aangepast om overmatige toename van het aantal bloedplaatjes tijdens de behandeling met eltrombopag te voorkomen.
Het is noodzakelijk om ten minste 2 weken te wachten om het effect van eventuele dosiswijzigingen op de trombocytenrespons van de patiënt te zien voordat een nieuwe dosisaanpassing wordt overwogen.
De standaard dosisaanpassing van eltrombopag, naar beneden of naar boven, dient 25 mg eenmaal daags te zijn.
Stopzetting van de behandeling
Behandeling met eltrombopag dient te worden gestaakt als het aantal bloedplaatjes niet stijgt tot een niveau dat voldoende is om klinisch belangrijke bloedingen te voorkomen na vier weken behandeling met 75 mg eltrombopag eenmaal daags.
Patiënten dienen een periodieke klinische evaluatie te ondergaan en de voortzetting van de behandeling dient op individuele basis door de arts te worden beslist. Bij patiënten die geen splenectomie hebben ondergaan, moet dit een evaluatie van de splenectomie omvatten. Herhaling van trombocytopenie is mogelijk na stopzetting van de behandeling (zie rubriek 4.4).
Trombocytopenie geassocieerd met chronische HCV-hepatitis
Wanneer eltrombopag wordt toegediend in combinatie met antivirale middelen, dient de samenvatting van de productkenmerken van de respectievelijke gelijktijdig toegediende geneesmiddelen te worden geraadpleegd voor volledige details van de relevante veiligheidsinformatie en contra-indicaties.
In klinische onderzoeken begon het aantal bloedplaatjes over het algemeen binnen 1 week na het starten met eltrombopag te stijgen. Het doel van de behandeling met eltrombopag moet zijn om het minimale aantal bloedplaatjes te bereiken dat nodig is om antivirale therapie te starten, in overeenstemming met de aanbevelingen in de klinische praktijk. het doel van de behandeling moet zijn het aantal bloedplaatjes op een niveau te houden dat het risico op bloedingscomplicaties voorkomt, meestal rond de 50.000 - 75.000 / l Het aantal bloedplaatjes > 75.000 / l moet worden vermeden De laagste dosis eltrombopag die nodig is om de doelen te bereiken, moet worden gebruikt Dosisaanpassingen zijn gebaseerd op de respons van het aantal bloedplaatjes.
Initiële doseringsschema
Eltrombopag moet worden gestart met een dosis van 25 mg eenmaal daags. Er is geen dosisaanpassing nodig voor patiënten met chronische HCV-hepatitis van Oost-Aziatische oorsprong of voor patiënten met een lichte leverfunctiestoornis (zie rubriek 5.2).
Dosisbewaking en -aanpassing
De dosis eltrombopag moet om de 2 weken worden gewijzigd in stappen van 25 mg om het beoogde aantal bloedplaatjes te bereiken dat nodig is om antivirale therapie te starten. Het aantal bloedplaatjes moet wekelijks worden gecontroleerd voordat met antivirale therapie wordt begonnen. Het aantal bloedplaatjes kan dalen bij het starten van een antivirale therapie, daarom moeten onmiddellijke veranderingen in de dosis eltrombopag worden vermeden (zie tabel 2).
Tijdens antivirale therapie moet de dosis eltrombopag zo nodig worden aangepast om verlagingen van de dosis peginterferon als gevolg van verlagingen van het aantal bloedplaatjes te voorkomen, waardoor de patiënt aan het risico op bloedingen zou kunnen worden blootgesteld (zie tabel 2). Het aantal bloedplaatjes moet wekelijks worden gecontroleerd tijdens antivirale therapie totdat een stabiel aantal bloedplaatjes is bereikt, meestal rond de 50.000-75.000 / microliter. Vervolgens dient maandelijks een volledige bloedtelling te worden uitgevoerd, inclusief het aantal bloedplaatjes en een uitstrijkje van perifeer bloed. Als het aantal bloedplaatjes de vereiste streefwaarde overschrijdt, moet een dosisverlaging van 25 mg van de dagelijkse dosis worden overwogen. Het is raadzaam om 2 weken te wachten om de effecten hiervan en eventuele daaropvolgende dosisaanpassingen te evalueren.
Een dosis van 100 mg eltrombopag eenmaal daags mag niet worden overschreden.
Tabel 2 Dosisaanpassingen van eltrombopag bij patiënten met chronische HCV-hepatitis tijdens antivirale therapie
* - Bij patiënten die eenmaal daags 25 mg eltrombopag innemen, moet worden overwogen om de behandeling met 25 mg om de andere dag te hervatten.
? - Het aantal bloedplaatjes kan afnemen bij het starten van een antivirale therapie, daarom moet een onmiddellijke verlaging van de dosis eltrombopag worden vermeden.
Stopzetting van de behandeling
Behandeling met eltrombopag dient te worden gestaakt als het aantal bloedplaatjes dat nodig is om antivirale therapie te starten niet is bereikt na 2 weken behandeling met 100 mg.
Tenzij anders gerechtvaardigd, moet de behandeling met eltrombopag worden stopgezet wanneer de antivirale therapie wordt stopgezet. Overmatige reacties op het aantal bloedplaatjes of ernstige afwijkingen in leverfunctietesten vereisen ook stopzetting van de behandeling.
Ernstige aplastische anemie
Initiële doseringsschema
De behandeling met eltrombopag moet beginnen met een dosis van 50 mg eenmaal daags. Voor patiënten van Oost-Aziatische afkomst dient de behandeling met eltrombopag te worden gestart met een verlaagde dosis van 25 mg eenmaal daags (zie rubriek 5.2). De behandeling mag niet worden gestart als patiënten reeds bestaande cytogenetische afwijkingen van chromosoom 7 hebben.
Dosisbewaking en -aanpassing
Voor hematologische respons is dosistitratie nodig, gewoonlijk tot 150 mg, en dit kan tot 16 weken duren na het starten van de behandeling met eltrombopag (zie rubriek 5.1) De dosis eltrombopag moet om de 2 weken worden veranderd om de streefwaarde te bereiken aantal bloedplaatjes ≥ 50.000 / l Voor patiënten die 25 mg eenmaal daags innemen, moet de dosis worden verhoogd tot 50 mg per dag voordat de volgende stappen van 50 mg worden genomen. Het mag niet worden overschreden. een dosis van 150 mg per dag Klinische hematologische en leveronderzoeken moeten worden uitgevoerd gecontroleerd tijdens de behandeling met eltrombopag en het aangepaste doseringsschema voor eltrombopag op basis van het aantal bloedplaatjes zoals aangegeven in Tabel 3.
Tabel 3 Dosisaanpassingen eltrombopag bij patiënten met ernstige aplastische anemie
Vermindering voor patiënten met trilineaire respons (witte bloedcellen, rode bloedcellen en bloedplaatjes)
Voor patiënten die een trilineaire respons bereiken, inclusief transfusieonafhankelijkheid, die ten minste 8 weken aanhoudt: de dosis eltrombopag kan met 50% worden verlaagd.
Als het bloedbeeld stabiel blijft na 8 weken bij de verlaagde dosis, moet de behandeling met eltrombopag worden gestaakt en moet het bloedbeeld worden gecontroleerd. Als het aantal bloedplaatjes zou dalen van een hemoglobineniveau naar een neutrofielenniveau naar een niveau
Onderbreking
Als er na 16 weken behandeling met eltrombopag geen hematologische respons is opgetreden, dient de behandeling te worden gestaakt. Indien nieuwe cytogenetische afwijkingen worden ontdekt, dient te worden overwogen of voortzetting van de behandeling met eltrombopag aangewezen is (zie rubrieken 4.4 en 4.8). Overmatige respons op het aantal bloedplaatjes (zoals aangegeven in Tabel 3) of ernstige afwijkingen in de levertest vereisen ook stopzetting van eltrombopag (zie rubriek 4.8).
Speciale populaties
Nierfalen
Er is geen dosisaanpassing nodig bij patiënten met nierinsufficiëntie. Patiënten met een verminderde nierfunctie dienen eltrombopag met voorzichtigheid en onder zorgvuldig toezicht te gebruiken, bijvoorbeeld door serumcreatinine en/of urineonderzoek uit te voeren (zie rubriek 5.2).
Leverinsufficiëntie
Eltrombopag mag niet worden gebruikt bij ITP-patiënten met leverinsufficiëntie (Child-Pugh-score ≥ 5), tenzij het verwachte voordeel opweegt tegen het vastgestelde risico op poortadertrombose (zie rubriek 4.4).
Als het gebruik van eltrombopag noodzakelijk wordt geacht voor ITP-patiënten met leverinsufficiëntie, dient de aanvangsdosis 25 mg eenmaal daags te zijn.Na aanvang van de toediening van de dosis eltrombopag bij patiënten met leverinsufficiëntie, dient een interval van 3 weken in acht te worden genomen voordat de dosis wordt verhoogd.
Er is geen dosisaanpassing nodig voor trombocytopenische patiënten met chronische HCV-hepatitis en milde leverinsufficiëntie (Child-Pugh-score ≤ 6). Patiënten met chronische HCV-hepatitis en ernstige aplastische anemie met leverinsufficiëntie dienen eltrombopag te starten met een dosis van 25 mg eenmaal daags (zie rubriek 5.2). Na het starten van de behandeling met eltrombopag bij patiënten met leverinsufficiëntie dient een interval van 2 weken in acht te worden genomen voordat de dosis wordt verhoogd.
Er is een verhoogd risico op bijwerkingen, waaronder leverdecompensatie en trombo-embolische voorvallen, bij trombocytopenische patiënten met gevorderde chronische leverziekte die worden behandeld met eltrombopag, hetzij ter voorbereiding op invasieve procedures, of bij patiënten met chronische HCV-hepatitis die worden behandeld met antivirale therapie (zie rubrieken 4.4 en 4.8).
Bejaarden
Er zijn beperkte gegevens over het gebruik van eltrombopag bij patiënten met ITP van 65 jaar en ouder en geen klinische ervaring bij patiënten met ITP ouder dan 85. In klinische onderzoeken met eltrombopag werden geen algemene klinische verschillen waargenomen. proefpersonen van ten minste 65 jaar en jongere proefpersonen Andere gerapporteerde klinische ervaringen hebben geen verschillen in respons tussen oudere en jongere patiënten aangetoond, maar een grotere gevoeligheid van sommige oudere proefpersonen kan niet worden uitgesloten (zie rubriek 5.2).
Er zijn beperkte gegevens over het gebruik van eltrombopag bij patiënten met chronische HCV-hepatitis en AAS ouder dan 75 jaar. Voorzichtigheid is geboden bij deze patiënten (zie rubriek 4.4).
Oost-Aziatische patiënten
Bij patiënten van Oost-Aziatische afkomst (zoals Chinees, Japans, Taiwanees, Koreaans of Thais), inclusief patiënten met een leverfunctiestoornis, dient eltrombopag te worden gestart met een dosis van 25 mg eenmaal daags (zie rubriek 5.2).
Het aantal bloedplaatjes van de patiënt moet worden gecontroleerd en de standaardcriteria voor verdere dosisaanpassingen moeten worden gevolgd.
Pediatrische populatie
Revolade wordt niet aanbevolen bij kinderen jonger dan 1 jaar met chronische ITP vanwege onvoldoende gegevens over veiligheid en werkzaamheid De veiligheid en werkzaamheid van eltrombopag bij kinderen en adolescenten (
Wijze van toediening
Oraal gebruik.
De tabletten moeten ten minste twee uur vóór of vier uur na een product worden ingenomen, zoals maagzuurremmers, zuivelproducten (of andere voedingsproducten die calcium bevatten) of minerale supplementen die polyvalente kationen bevatten (bijv. ijzer, calcium, magnesium, aluminium, selenium en zink). ) (zie rubrieken 4.5 en 5.2).
04.3 Contra-indicaties
Overgevoeligheid voor eltrombopag of voor één van de in rubriek 6.1 vermelde hulpstoffen.
04.4 Bijzondere waarschuwingen en passende voorzorgen bij gebruik
Er is een verhoogd risico op bijwerkingen, waaronder levensbedreigende leverdecompensatie en trombo-embolische voorvallen, bij patiënten met trombocytopenische HCV chronische hepatitis met gevorderde chronische leverziekte, gedefinieerd door een laag albumine ≤ 35 g/l of door een Model voor score. Ziekte (MELD) ≥ 10, indien behandeld met eltrombopag in combinatie met op interferon gebaseerde therapie. Bovendien waren de voordelen van de behandeling in termen van het percentage patiënten dat aanhoudende virologische respons (SVR) bereikte in vergelijking met placebo bescheiden bij deze patiënten (vooral die met een baseline albumine ≤ 35 g/l) in vergelijking met de totale groep. Behandeling met eltrombopag bij deze patiënten mag alleen worden gestart door artsen met ervaring in de behandeling van gevorderde chronische HCV-hepatitis, en alleen wanneer het risico op trombocytopenie of stopzetting van antivirale therapie interventie vereist. Als de behandeling als klinisch geïndiceerd wordt beschouwd, is nauwlettende monitoring van deze patiënten vereist.
Combinatie met direct werkende antivirale middelen
De veiligheid en werkzaamheid zijn niet vastgesteld in combinatie met direct werkende antivirale middelen die zijn goedgekeurd voor de behandeling van chronische HCV-hepatitis.
Risico op hepatotoxiciteit
Toediening van eltrombopag kan een abnormale leverfunctie en ernstige hepatotoxiciteit veroorzaken, die levensbedreigend kan zijn. In gecontroleerde klinische onderzoeken met eltrombopag bij chronische ITP werden verhogingen van serum-alanine-aminotransferase (ALAT), aspartaat-aminotransferase (ASAT) en bilirubine waargenomen (zie rubriek 4.8).
Deze veranderingen waren meestal mild (graad 1-2), reversibel en gingen niet gepaard met klinisch significante symptomen die op een "verminderde leverfunctie" zouden hebben geduid. In" de 3 placebogecontroleerde onderzoeken bij volwassenen met chronische ITP, 1 patiënt van de placebogroep en 1 patiënt van de eltrombopag-groep had een graad 4 afwijking in leverfunctieparameters. In twee placebogecontroleerde onderzoeken bij pediatrische patiënten (in de leeftijd van 1 tot 17 jaar) met chronische ITP, een ALT-waarde ≥ 3 maal de bovengrens van normaal ( x ULN) werd gezien bij respectievelijk 4,7% en 0% van de eltrombopag- en placebogroepen.
In 2 gecontroleerde klinische onderzoeken bij patiënten met chronische HCV-hepatitis werden ALT of ASAT ≥ 3 maal de bovengrens van normaal (ULN) gemeld bij respectievelijk 34% en 38% van de eltrombopag- en placebogroepen. De meeste patiënten die eltrombopag hebben gekregen in combinatie met behandeling met peginterferon/ribavirine zullen indirecte hyperbilirubinemie ervaren. In totaal werd totaal bilirubine ≥ 1,5 maal ULN gemeld in respectievelijk 76% en 50% van de eltrombopag- en placebogroepen.
Serum-ALAT, ASAT en serumbilirubine moeten worden gemeten voordat met eltrombopag wordt begonnen, elke 2 weken tijdens de dosisaanpassingsfase en maandelijks na het bereiken van een stabiele dosis.
Eltrombopag remt UDP glucorosyltransferase (UGT) 1A1 en organisch aniontransporterpolypeptide (OATP) 1B1, wat kan leiden tot indirecte hyperbilirubinemie. Fractionering moet worden uitgevoerd als bilirubine verhoogd is. Afwijkingen in serumleverfunctietests moeten worden beoordeeld door binnen 3-5 opnieuw te testen dagen Als afwijkingen worden bevestigd, moeten serumleverfunctietests worden gecontroleerd totdat de afwijkingen zijn verdwenen, gestabiliseerd of terugkeren naar de uitgangswaarde.
De toediening van eltrombopag dient te worden gestaakt als de ALT-spiegels stijgen (≥ 3 maal de ULN bij patiënten met een normale leverfunctie, of ≥ 3 maal de uitgangswaarde of > 5 maal de ULN, welke van beide lager is, bij patiënten met verhogingen van de transaminasen vóór de behandeling) en zijn:
- progressief, of
- 4 weken aanhouden, of
- gepaard gaan met een toename van direct bilirubine, of
- gepaard gaan met klinische symptomen van leverbeschadiging of tekenen van leverdecompensatie.
Voorzichtigheid is geboden bij toediening van eltrombopag aan patiënten met een leveraandoening. Bij patiënten met ITP en SAA moet een lagere startdosis eltrombopag worden gebruikt. Bij toediening aan patiënten met leverinsufficiëntie is zorgvuldige controle vereist (zie rubriek 4.2).
Leverfalen (gebruik met interferon)
Leverdecompensatie bij patiënten met chronische HCV-hepatitis: Controle is vereist bij patiënten met lage albuminespiegels (≤ 35 g/l) of met een baseline MELD-score ≥ 10.
Patiënten met chronische HCV-hepatitis en cirrose kunnen een risico lopen op leverdecompensatie wanneer zij behandeld worden met alfa-interferon. In 2 gecontroleerde klinische onderzoeken bij trombocytopenische patiënten met chronische HCV-hepatitis werd leverdecompensatie (ascites, hepatische encefalopathie, varicesbloeding, spontane bacteriële peritonitis) vaker gemeld in de eltrombopag-arm (11%) dan in de placebo-arm (6%). Bij patiënten met lage albuminespiegels (≤ 35 g/l) of MELD-score ≥ 10 bij baseline, was er een drievoudig verhoogd risico op leverdecompensatie en een verhoogd risico op fatale bijwerkingen in vergelijking met patiënten met een minder gevorderde lever. Verder waren de behandelingsvoordelen in termen van mate van realisatie van SVR vergeleken met placebo bescheiden bij deze patiënten (vooral die met baseline albumine ≤ 35g/l) in vergelijking met de totale groep. Eltrombopag mag alleen aan deze patiënten worden toegediend na zorgvuldige afweging van de verwachte voordelen versus de risico's. Patiënten met deze kenmerken moeten zorgvuldig worden gecontroleerd op tekenen en symptomen van leverdecompensatie. Voor stopzettingscriteria dient te worden verwezen naar de respectievelijke samenvatting van de productkenmerken van interferon. Eltrombopag moet worden stopgezet als antivirale therapie wordt stopgezet vanwege leverdecompensatie.
Trombotische / trombo-embolische complicaties
In gecontroleerde klinische onderzoeken bij trombocytopenische patiënten met chronische HCV-hepatitis die op interferon gebaseerde therapie kregen (n = 1439), vertoonden 38 van de 955 (4%) proefpersonen die werden behandeld met eltrombopag en 6 van de 484 (1%) proefpersonen in de placebogroep trombo-embolische voorvallen (TEE). Meldingen van trombotische/trombo-embolische complicaties omvatten zowel veneuze als arteriële voorvallen. De meeste TEE's waren niet-ernstig en waren aan het einde van het onderzoek opgelost. Poortadertrombose was de meest voorkomende TEE in beide behandelingsgroepen (2% bij patiënten behandeld met eltrombopag vergeleken met tekenen en symptomen van TEE.
Het risico op TEE was verhoogd bij patiënten met chronische leverziekte (chronische leverziekte, CLD) behandeld met 75 mg eltrombopag eenmaal daags gedurende twee weken ter voorbereiding op invasieve procedures.
Zes van de 143 (4%) volwassen patiënten met CLD die eltrombopag kregen, kregen TEE's (allemaal met betrekking tot het veneuze portale systeem) en twee van de 145 proefpersonen (1%) in de placebogroep kregen TEE's (één met betrekking tot het veneuze portale systeem en een myocardinfarct) . Vijf van de 6 met eltrombopag behandelde patiënten ondervonden trombotische complicaties met een trombocytenaantal > 200.000/microliter en binnen 30 dagen na de laatste dosis eltrombopag Eltrombopag is niet geïndiceerd voor de behandeling van trombocytopenie bij patiënten met chronische leverziekte ter voorbereiding op invasieve procedures .
In klinische onderzoeken met eltrombopag bij ITP werden trombo-embolische voorvallen waargenomen met een laag en normaal aantal bloedplaatjes. Voorzichtigheid is geboden bij het toedienen van eltrombopag aan patiënten met bekende risicofactoren voor trombo-embolie, inclusief maar niet beperkt tot erfelijke (bijv. Factor V Leiden) of verworven (bijv. ATIII-deficiëntie, antifosfolipidensyndroom) risicofactoren, oudere leeftijd, patiënten met langdurige perioden van immobilisatie , maligniteiten, anticonceptie of hormoonvervangende therapie, chirurgie/trauma, zwaarlijvigheid en roken. Het aantal bloedplaatjes moet nauwlettend worden gecontroleerd en dosisverlaging of stopzetting van eltrombopag moet worden overwogen als het aantal bloedplaatjes de vereiste niveaus overschrijdt (zie rubriek 4.2.) Bij patiënten met een risico op TEE-gebeurtenissen van welke etiologie dan ook, moet rekening worden gehouden met de baten-risicoverhouding.
Eltrombopag mag niet worden gebruikt bij ITP-patiënten met leverinsufficiëntie (Child-Pugh-score ≥ 5), tenzij het verwachte voordeel opweegt tegen het vastgestelde risico op poortadertrombose. Wanneer behandeling geschikt wordt geacht, is voorzichtigheid geboden als eltrombopag wordt toegediend aan patiënten met leverinsufficiëntie (zie rubrieken 4.2 en 4.8).
Bloeding na stopzetting van eltrombopag
Trombocytopenie zal waarschijnlijk opnieuw optreden na stopzetting van de behandeling met eltrombopag. Na stopzetting van de behandeling met eltrombopag keert het aantal bloedplaatjes bij de meeste patiënten binnen 2 weken terug naar de uitgangswaarde, wat het risico op bloedingen verhoogt en in sommige gevallen kan leiden tot bloedingen.Dit risico neemt toe als de behandeling met eltrombopag wordt stopgezet in aanwezigheid van anticoagulantia en bloedplaatjesmiddelen Het wordt aanbevolen om de ITP-behandeling te hervatten volgens de huidige richtlijnen als de behandeling met eltrombopag wordt stopgezet. Daarnaast kan de medische behandeling bestaan uit het staken van anticoagulantia en/of anticoagulatietherapie - bloedplaatjes, omkering van de antistolling of bloedplaatjesondersteuning. tellingen dienen wekelijks te worden gecontroleerd gedurende 4 weken na het stoppen met eltrombopag.
In klinische onderzoeken naar chronische HCV-hepatitis is een hogere incidentie van maagbloeding gemeld, waaronder ernstige en fatale gevallen, na stopzetting van peginterferon, ribavirine en eltrombopag.
Na stopzetting van de behandeling moeten patiënten worden gecontroleerd op tekenen of symptomen van maagbloeding.
Vorming van reticuline in het beenmerg en risico op beenmergfibrose Eltrombopag kan het risico op ontwikkeling of progressie van reticulinevezels in het beenmerg verhogen.Net als bij andere trombopoëtinereceptoragonisten (TPO-R), is de relevantie van deze veranderingen nog niet vastgesteld.
Voordat met eltrombopag wordt gestart, moet het uitstrijkje van perifeer bloed zorgvuldig worden onderzocht om het baselineniveau van cellulaire morfologische afwijkingen vast te stellen. Na identificatie van een stabiele dosis eltrombopag moet maandelijks een volledige bloedtelling met differentiële witte bloedceltelling worden uitgevoerd.Als onrijpe of dysplastische cellen worden waargenomen, moet een perifeer bloeduitstrijkje worden onderzocht op nieuwe morfologische afwijkingen (bijvoorbeeld traanvormige rode bloedcellen (dacryocyten) en genucleëerde, onvolgroeide witte bloedcellen) of verergering of cytopenie Als de patiënt nieuwe of verergerende morfologische afwijkingen of cytopenie ontwikkelt, moet de behandeling met eltrombopag worden stopgezet en genomen. Overweeg een beenmergbiopsie, inclusief evaluatie van fibrose.
Progressie van bestaand myelodysplastisch syndroom (MDS)
TPO-R-agonisten zijn groeifactoren die proliferatie en differentiatie van trombopoëtische voorlopercellen en de productie van bloedplaatjes induceren. TPO-R wordt voornamelijk tot expressie gebracht op het oppervlak van myeloïde afstammingscellen. Voor TPO-R-agonisten bestaat het risico dat ze de progressie van reeds bestaande neoplastische hemopathieën zoals myelodysplastisch syndroom kunnen stimuleren.
In klinische onderzoeken met een TPO-R-agonist bij patiënten met MDS zijn gevallen van voorbijgaande toename van het aantal blastcellen waargenomen en zijn gevallen van ziekteprogressie van MDS naar acute myeloïde leukemie (AML) gemeld.
De diagnose ITP of SAA bij volwassen en oudere patiënten moet worden bevestigd door andere pathologieën die trombocytopenie vertonen uit te sluiten, met name de diagnose MDS moet worden uitgesloten. Beenmergaspiratie en biopsie moeten worden overwogen tijdens de ziekte en de behandeling, vooral bij patiënten ouder dan 60 jaar met systemische symptomen of abnormale verschijnselen zoals een toename van perifere blastcellen.
De werkzaamheid en veiligheid van eltrombopag zijn niet vastgesteld voor gebruik bij andere trombocytopenische aandoeningen, waaronder door chemotherapie geïnduceerde trombocytopenie of MDS.
Eltrombopag mag niet worden gebruikt buiten klinische onderzoeken voor de behandeling van trombocytopenie als gevolg van MDS of enige andere oorzaak van trombocytopenie anders dan de goedgekeurde indicaties.
Cytogenetische afwijkingen en progressie van MDS/AML bij patiënten met AAS
Het is bekend dat cytogenetische afwijkingen optreden bij patiënten met AAS. Het is niet bekend of eltrombopag het risico op cytogenetische afwijkingen bij patiënten met AAS verhoogt. In de klinische fase II-studie waarin eltrombopag werd gebruikt bij AAS, werd de incidentie van nieuwe cytogenetische afwijkingen waargenomen bij 19% van de patiënten [8/43 (waarvan 5 met chromosoom 7-afwijkingen)]. De mediane tijd tijdens de studie voor het optreden van een cytogenetische afwijking was 2,9 maanden.
In klinische onderzoeken met eltrombopag bij ASA werd bij 4% van de patiënten (5/133) MDS vastgesteld.De mediane tijd tot diagnose vanaf de start van de behandeling met eltrombopag was drie maanden.
Voor patiënten met refractaire of zwaar voorbehandelde SAA en die een eerdere immunosuppressieve therapie ondergaan, wordt beenmergaspiraatonderzoek op cytogenetica aanbevolen voordat met eltrombopag wordt begonnen, na 3 maanden behandeling en daarna om de 6 maanden. In geval van detectie van nieuwe cytogenetische afwijkingen, dient dit moet worden overwogen of het passend is om door te gaan met eltrombopag.
Oculaire veranderingen
Cataract is waargenomen in eltrombopag-toxicologische onderzoeken bij knaagdieren (zie rubriek 5.3). In gecontroleerde klinische onderzoeken bij trombocytopenische patiënten met chronische HCV-hepatitis die interferontherapie kregen (n = 1439), werd progressie van een reeds bestaande baseline cataract of het verschijnen van een nieuwe cataract gemeld bij 8% van de groep eltrombopag en bij 5% van de Retinale bloedingen, voornamelijk graad 1 of 2, werden gemeld bij patiënten met chronische HCV-hepatitis die interferon, ribavirine en eltrombopag kregen (2% in de eltrombopag-groep en 2% in de placebogroep). netvlies (preretinaal), onder het netvlies (subretinaal) of in het netvliesweefsel. Routine oogheelkundige controle van patiënten wordt aanbevolen.
QT / QTc-extensie
Een QTc-onderzoek bij gezonde vrijwilligers met een dosis van 150 mg eltrombopag per dag toonde geen klinisch significant effect op cardiale repolarisatie. Verlenging van het QTc-interval is gemeld in klinische onderzoeken met ITP-patiënten en trombocytopenische patiënten met chronische HCV-hepatitis. De klinische betekenis van deze gevallen van QTc-verlenging is niet bekend.
Verlies van respons op eltrombopag
Verlies van respons of het niet handhaven van de bloedplaatjesrespons op behandeling met eltrombopag binnen het aanbevolen therapeutische bereik zou moeten leiden tot het zoeken naar oorzakelijke factoren, waaronder een toename van beenmergreticuline.
Pediatrische populatie
De hierboven vermelde ITP-waarschuwingen en voorzorgsmaatregelen worden ook toegepast op de pediatrische populatie.
04.5 Interacties met andere geneesmiddelen en andere vormen van interactie
Effecten van eltrombopag op andere geneesmiddelen
HMG CoA-reductaseremmers
Opleiding in vitro aangetoond dat eltrombopag geen substraat is van het organische aniontransporterpolypeptide OATP1B1, maar een remmer van deze transporter is. Opleiding in vitro hebben ook aangetoond dat eltrombopag een substraat en een remmer is van het borstkankerresistentie-eiwit (BCRP). Toediening aan 39 gezonde volwassen proefpersonen van 75 mg eltrombopag eenmaal daags gedurende 5 dagen met een enkelvoudige dosis van 10 mg rosuvastatine, een substraat van OATP1B1 en BCRP, verhoogde de plasma-Cmax van rosuvastatine met 103% (90% betrouwbaarheidsinterval [BI]: 82 %, 126%) en de AUC0-? 55% (90%-BI: 42%, 69%). Interacties met andere HMG-CoA-reductaseremmers worden ook verwacht, waaronder atorvastatine, fluvastatine, lovastatine, pravastatine en simvastatine. Bij gelijktijdige toediening met eltrombopag moet een dosisverlaging van statines worden overwogen en moet zorgvuldig worden gecontroleerd op bijwerkingen van statines (zie rubriek 5.2).
OATP1B1- en BCRP-substraten
Gelijktijdige toediening van eltrombopag en substraten van OATP1B1 (bijv. methotrexaat) en BCRP (bijv. topotecan en methotrexaat) dient met voorzichtigheid te gebeuren (zie rubriek 5.2).
Cytochroom P450-substraten
In onderzoeken met humane levermicrosomen vertoonde eltrombopag (tot 100 mcM) geen remming. in vitro van CYP450-enzymen 1A2, 2A6, 2C19, 2D6, 2E1, 3A4/5 en 4A9/11 en was een remmer van CYP2C8 en CYP2C9, gemeten met paclitaxel en diclofenac als probesubstraten. Toediening van 75 mg eltrombopag eenmaal daags gedurende 7 dagen aan 24 gezonde mannelijke proefpersonen remde of induceerde het metabolisme van probesubstraten voor 1A2 (cafeïne), 2C19 (omeprazol), 2C9 (flurbiprofen) of 3A4 (midazolam) bij de "mens. Er worden geen klinisch significante interacties verwacht wanneer eltrombopag en CYP450-substraten gelijktijdig worden toegediend (zie rubriek 5.2).
HCV-proteaseremmers
Er is geen dosisaanpassing nodig wanneer eltrombopag gelijktijdig wordt toegediend met telaprevir of boceprevir.
Gelijktijdige toediening van een enkele dosis eltrombopag 200 mg met telaprevir 750 mg om de 8 uur veranderde de plasmablootstelling van telaprevir niet.
Gelijktijdige toediening van een enkele dosis eltrombopag 200 mg met boceprevir 800 mg om de 8 uur veranderde de plasma-AUC (0-?) van boceprevir niet, maar verhoogde de Cmax met 20% en verlaagde de Cmin met 32%. verlaging van de Cmin is niet vastgesteld: nauwkeurigere klinische en laboratoriumcontrole wordt aanbevolen voor onderdrukking van HCV.
Effecten van andere geneesmiddelen op eltrombopag
Cyclosporine
In vitro-onderzoeken hebben aangetoond dat eltrombopag een substraat en remmer van BCRP is. Een verlaging van de blootstelling aan eltrombopag werd waargenomen bij gelijktijdige toediening van 200 mg en 600 mg ciclosporine (BCRP-remmer) (zie rubriek 5.2) Dosisaanpassing van eltrombopag tijdens de behandeling is toegestaan op basis van het aantal bloedplaatjes van de patiënt (zie rubriek 4.2). Bij gelijktijdige toediening van eltrombopag met ciclosporine moet het aantal bloedplaatjes ten minste wekelijks worden gecontroleerd gedurende 2 tot 3 weken. De dosis eltrombopag moet mogelijk worden verhoogd op basis van de resultaten van het aantal bloedplaatjes.
Polyvalente kationen (chelatie)
Eltrombopag cheleert polyvalente kationen zoals ijzer, calcium, magnesium, aluminium, selenium en zink. Toediening van een enkelvoudige dosis van 75 mg eltrombopag met een antacidum dat een polyvalent kation bevat (1524 mg aluminiumhydroxide en 1425 mg magnesiumcarbonaat) verlaagt de AUC0-? plasma-eltrombopag tot 70% (90% BI: 64%, 76%) en Cmax tot 70% (90% BI: 62%, 76%).
Eltrombopag moet ten minste 2 uur vóór of 4 uur na elk antacidum-type product, zuivelproduct of mineraalsupplement dat polyvalente kationen bevat, worden ingenomen om een significante vermindering van de eltrombopag-absorptie als gevolg van chelatie te voorkomen (zie rubrieken 4.2 en 5.2).
Interactie met voedsel
Toediening van eltrombopag-tabletten of poeder voor orale suspensie met een calciumrijke maaltijd (bijv. een maaltijd met zuivelproducten) verminderde de AUC0-? en plasma Cmax van eltrombopag. Omgekeerd toediening van eltrombopag 2 uur vóór of 4 uur na een calciumrijke maaltijd of calciumarm voedsel [
Lopinavir / ritonavir
Gelijktijdige toediening van eltrombopag met lopinavir/ritonavir kan een verlaging van de eltrombopagconcentratie veroorzaken. Een onderzoek bij 40 gezonde vrijwilligers toonde aan dat gelijktijdige toediening van een enkelvoudige dosis van 100 mg eltrombopag met een herhaalde dosis van 400/100 mg lopinavir/ritonavir tweemaal daags resulteerde in een verlaging van de AUC (0-?) van eltrombopag van 17% (90 % BI: 6,6%; 26,6%). Daarom is voorzichtigheid geboden bij gelijktijdige toediening van eltrombopag met lopinavir/ritonavir. Het aantal bloedplaatjes moet nauwlettend worden gecontroleerd om zeker te zijn van een geschikt klinisch dosisbeheer van eltrombopag wanneer de behandeling met lopinavir/ritonavir wordt gestart of stopgezet.
CYP1A2- en CYP2C8-remmers en -inductoren
Eltrombopag wordt gemetaboliseerd via meerdere routes, waaronder CYP1A2, CYP2C8, UGT1A1 en UGT1A3 (zie rubriek 5.2). Geneesmiddelen die een enkel enzym remmen of induceren, hebben waarschijnlijk geen significante invloed op de plasmaconcentraties van eltrombopag, terwijl geneesmiddelen die meerdere enzymen remmen of induceren het potentieel hebben om de concentraties van eltrombopag te verhogen (bijv. fluvoxamine) of te verlagen (bijv. rifampicine).
HCV-proteaseremmers
De resultaten van een farmacokinetisch onderzoek naar geneesmiddelinteracties tonen aan dat gelijktijdige toediening van herhaalde doses boceprevir 800 mg om de 8 uur of telaprevir 750 mg om de 8 uur met een enkele dosis eltrombopag 200 mg de plasmablootstelling aan eltrombopag niet veranderde. klinisch significante niveaus.
Geneesmiddelen voor de behandeling van ITP
Geneesmiddelen die in klinische onderzoeken bij de behandeling van ITP in combinatie met eltrombopag werden gebruikt, waren onder meer corticosteroïden, danazol en/of azathioprine, intraveneuze immunoglobuline (IVIG) en anti-D-immunoglobuline. Het aantal bloedplaatjes moet worden gecontroleerd wanneer eltrombopag wordt gegeven in combinatie met andere geneesmiddelen voor de behandeling van ITP, om te voorkomen dat het aantal bloedplaatjes buiten het aanbevolen bereik valt (zie rubriek 4.2).
04.6 Zwangerschap en borstvoeding
Zwangerschap
Er zijn geen of een beperkte hoeveelheid gegevens over het gebruik van eltrombopag bij zwangere vrouwen Dierstudies hebben reproductietoxiciteit aangetoond (zie rubriek 5.3). Het potentiële risico voor mensen is niet bekend.
Revolade wordt niet aanbevolen tijdens de zwangerschap.
Vrouwen in de vruchtbare leeftijd / Anticonceptie bij mannen en vrouwen
Revolade wordt niet aanbevolen voor vrouwen in de vruchtbare leeftijd die geen voorbehoedsmiddelen gebruiken.
Voedertijd
Het is niet bekend of eltrombopag/zijn metabolieten in de moedermelk worden uitgescheiden. Dierstudies hebben aangetoond dat eltrombopag waarschijnlijk wordt uitgescheiden in de melk (zie rubriek 5.3); daarom kan een risico voor de zuigeling niet worden uitgesloten. Er moet worden besloten of de borstvoeding moet worden gestaakt of dat de behandeling met Revolade moet worden voortgezet/afgezien, waarbij het voordeel moet worden geëvalueerd. van borstvoeding voor de baby en het voordeel van therapie voor de vrouw.
Vruchtbaarheid
De vruchtbaarheid werd niet beïnvloed bij mannelijke en vrouwelijke ratten bij blootstellingen die vergelijkbaar waren met die bij mensen, maar een risico voor mensen kan niet worden uitgesloten (zie rubriek 5.3).
04.7 Beïnvloeding van de rijvaardigheid en het vermogen om machines te bedienen
Eltrombopag heeft een verwaarloosbare invloed op de rijvaardigheid of op het vermogen om machines te bedienen. De klinische toestand van de patiënt en het bijwerkingenprofiel van eltrombopag, waaronder duizeligheid en gebrek aan alertheid, moeten in gedachten worden gehouden bij het overwegen van het vermogen van de patiënt om taken uit te voeren die beoordelingsvermogen, motorische en cognitieve vaardigheden vereisen.
04.8 Bijwerkingen
Samenvatting van het veiligheidsprofiel
In 4 gecontroleerde en 2 ongecontroleerde klinische onderzoeken werden 530 volwassen patiënten met chronische ITP behandeld met eltrombopag%. De gemiddelde duur van de blootstelling aan eltrombopag was 260 dagen De belangrijkste ernstige bijwerkingen waren hepatotoxiciteit en trombotische/trombo-embolische voorvallen. De meest voorkomende bijwerkingen die optraden bij ten minste 10% van de patiënten waren: hoofdpijn, bloedarmoede, verminderde eetlust, slapeloosheid, hoesten, misselijkheid, diarree, alopecia, pruritus, spierpijn, koorts, vermoeidheid, griepachtige ziekte, asthenie, koude rillingen en perifeer oedeem.
In 2 gecontroleerde klinische onderzoeken werden 171 pediatrische patiënten met chronische ITP behandeld met eltrombopag. De mediane blootstellingsduur was 171 dagen. Het bijwerkingenprofiel was vergelijkbaar met dat bij volwassenen met enkele aanvullende bijwerkingen, aangegeven in de volgende tabel.
De meest voorkomende bijwerkingen bij pediatrische patiënten van 1 jaar en ouder met ITP (≥ 3% en meer dan placebo) waren ernstige luchtweginfecties, nasofaryngitis, hoesten, diarree, koorts, rhinitis, buikpijn, orofaryngeale pijn, tandpijn, huiduitslag, verhoogde ASAT en rinorroe.
In 2 gecontroleerde klinische onderzoeken werden 955 trombocytopenische patiënten met HCV-infectie behandeld met eltrombopag. De mediane duur van blootstelling was 183 dagen. De belangrijkste ernstige bijwerkingen die werden vastgesteld waren hepatotoxiciteit en trombotische/trombo-embolische voorvallen. De meest voorkomende bijwerkingen die optraden bij ten minste 10% van de patiënten waren: hoofdpijn, anemie, verminderde eetlust, slapeloosheid, hoesten , misselijkheid, diarree, alopecia, jeuk, spierpijn, koorts, vermoeidheid, griepachtige ziekte, asthenie, koude rillingen en perifeer oedeem.
De veiligheid van eltrombopag bij ernstige aplastische anemie werd geëvalueerd in een open-label, eenarmige klinische studie (N = 43) waarin 12 patiënten (28%) werden behandeld gedurende > 6 maanden en 9 patiënten (21%) werden behandeld. > 1 jaar De belangrijkste ernstige bijwerkingen waren febriele neutropenie en sepsis/infecties De meest voorkomende bijwerkingen die optraden (bij ten minste 10% van de patiënten) waren: hoofdpijn, duizeligheid, slapeloosheid, hoesten, kortademigheid, orofaryngeale pijn, rinorroe, misselijkheid, diarree, buikpijn, verhoogde transaminasen, blauwe plekken, artralgie, spierspasmen, pijn in ledematen, vermoeidheid, febriele neutropenie en pyrexie.
Lijst met bijwerkingen
Bijwerkingen in ITP-onderzoeken bij volwassenen (N = 550), pediatrische ITP-onderzoeken (N = 107) en HCV-geïnfecteerde onderzoeken (N = 955), AAS-onderzoeken (N = 43) en postmarketingmeldingen worden hieronder vermeld per MedDRA-systeem/orgaanklasse en frequentie .
Zeer vaak (≥ 1/10)
Vaak (≥ 1/100 tot
Soms (≥ 1 / 1.000 tot
Zeldzaam (≥ 1 / 10.000 tot
Erg zeldzaam (
Niet bekend (kan met de beschikbare gegevens niet worden bepaald)
Populatie van de klinische proef in ITP
Infecties en parasitaire aandoeningen
Heel gewoon
Nasofaryngitis?, bovenste luchtweginfecties?
gemeenschappelijk
Rhinitis?
Ongewoon
Faryngitis, urineweginfecties, griep, orale herpes, longontsteking, sinusitis, tonsillitis, luchtweginfecties, gingivitis, huidinfectie
Neoplasmata, goedaardig, kwaadaardig en niet gespecificeerd (inclusief cysten en poliepen)
Ongewoon
Tumor van het rectosigmoïde kanaal
Aandoeningen van het bloed en het lymfestelsel
Ongewoon
Bloedarmoede, anisocytose, eosinofilie, hemolytische anemie, leukocytose, myelocytose, trombocytopenie, verhoogd hemoglobine, verhoogd aantal neutrofielen in de band, verlaagd hemoglobine, aanwezigheid van myelocyten, verhoogd aantal bloedplaatjes, verlaagd aantal witte bloedcellen.
Aandoeningen van het immuunsysteem
Ongewoon
overgevoeligheid
Metabolisme en voedingsstoornissen
Ongewoon
Anorexie, hypokaliëmie, verminderde eetlust, jicht, hypocalciëmie, verhoogd urinezuur in het bloed
Psychische stoornissen
Ongewoon
Slaapstoornissen, depressie, apathie, stemmingswisselingen, gemakkelijk huilen
Zenuwstelselaandoeningen
gemeenschappelijk
Paresthesieën
Ongewoon
Hypo-esthesie, slaperigheid, migraine, tremoren, evenwichtsstoornissen, dysesthesie, hemiparese, migraine met aura, perifere neuropathie, perifere sensorische neuropathie, spraakstoornissen, toxische neuropathie, vasculaire hoofdpijn
Oogaandoeningen
gemeenschappelijk
Droge ogen
Ongewoon
Wazig zien, lenticulaire opaciteit, astigmatisme, corticale cataract, oogpijn, verhoogde traanproductie, retinale bloeding, retinale pigmentepitheliopathie, verminderde gezichtsscherpte, verminderde gezichtsscherpte, afwijkingen van de gezichtsscherptetest, blefaritis en keratoconjunctivitis sicca
Oor- en labyrintaandoeningen
Ongewoon
Oorpijn, duizeligheid
Cardiale pathologieën
Ongewoon
Tachycardie, acuut myocardinfarct, cardiovasculaire aandoeningen, cyanose, sinustachycardie, QT-verlenging van het elektrocardiogram
Vasculaire pathologieën
Ongewoon
Diepe veneuze trombose, embolie, blozen, oppervlakkige tromboflebitis, roodheid, hematoom
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen
gemeenschappelijk
Hoesten?, orofaryngeale pijn?, rinorroe?
Ongewoon
Longembolie, longinfarct, ongemak in de neus, blaarvorming in de orofarynx, pijn in de orofarynx, sinusstoornissen, slaapapneusyndroom
Maagdarmstelselaandoeningen
gemeenschappelijk
Misselijkheid, diarree*, mondzweren, kiespijn?
* Zeer vaak bij pediatrische ITP-patiënten
Ongewoon
Droge mond, braken, buikpijn, glossodynie, bloeding in de mond, abdominale spanning, verkleurde ontlasting, winderigheid, voedselvergiftiging, frequente buikbewegingen, haematemesis, ongemak in de mond
Lever- en galaandoeningen
gemeenschappelijk
Alanineaminotransferase * verhoogd, aspartaataminotransferase * verhoogd, hyperbilirubinemie, leverfunctiestoornissen
Ongewoon
Cholestase, leverbeschadiging, hepatitis, door geneesmiddelen geïnduceerde leverbeschadiging
* Verhoging van alanineaminotransferase en aspartaataminotransferase kan gelijktijdig optreden, zij het met een lagere frequentie.
Huid- en onderhuidaandoeningen
gemeenschappelijk
Huiduitslag, alopecia
Ongewoon Hyperhidrose, gegeneraliseerde jeuk, urticaria, dermatose, petechiën, koud zweet, erytheem, melanose, pigmentatiestoornissen, huidverkleuring, vervellen van de huid
Skeletspierstelsel- en bindweefselaandoeningen
gemeenschappelijk
Myalgie, spierspasmen, musculoskeletale pijn, botpijn, rugpijn
Ongewoon
Spier zwakte
Nier- en urinewegaandoeningen
Ongewoon
Nierfalen, leukocyturie, lupoide nefritis, nocturie, proteïnurie, bloedureum verhoogd, bloedcreatinine verhoogd, eiwit/creatinineverhouding verhoogd
Ziekten van het voortplantingssysteem en de borst
gemeenschappelijk
menorragie
Algemene aandoeningen en toedieningsplaatsstoornissen
gemeenschappelijk
Pyrexie?
Ongewoon
Pijn op de borst, warm gevoel, bloeding op de parenterale injectieplaats, asthenie, zenuwachtig gevoel, wondontsteking, malaise, pyrexie, gevoel van vreemd lichaam
Diagnostische toetsen
Ongewoon
Verhoogd bloedalbumine, verhoogd bloed alkalische fosfatase, verhoogd totaal eiwit, verlaagd bloedalbumine, verhoogde urine-pH
Letsel, vergiftiging en procedurele complicaties
Ongewoon
Zonnebrand
? Bijkomende bijwerkingen waargenomen in pediatrische populatieonderzoeken (1 tot 17 jaar)
Met HCV geïnfecteerde klinische onderzoekspopulatie (in combinatie met antivirale therapie met interferon en ribavirine)
Infecties en parasitaire aandoeningen
gemeenschappelijk
Urineweginfecties, bovenste luchtweginfecties, bronchitis, nasofaryngitis, griep, orale herpes, gastro-enteritis, faryngitis.
Neoplasmata, goedaardig, kwaadaardig en niet gespecificeerd (inclusief cysten en poliepen)
gemeenschappelijk
Kwaadaardige levertumor
Aandoeningen van het bloed en het lymfestelsel
Heel gewoon
Bloedarmoede
gemeenschappelijk lymfocytopenie, hemolytische anemie
Metabolisme en voedingsstoornissen
Heel gewoon
Verminderde eetlust
gemeenschappelijk
Hyperglykemie, abnormaal gewichtsverlies
Psychische stoornissen
Heel gewoon
Slapeloosheid
gemeenschappelijk
Depressie, angst, slaapstoornissen, verwardheid, opwinding
Zenuwstelselaandoeningen
Heel gewoon
Hoofdpijn
gemeenschappelijk
Duizeligheid, aandachtsstoornis, dysgeusie, hepatische encefalopathie, lethargie, geheugenstoornis, paresthesie
Oogaandoeningen
gemeenschappelijk
Cataract, retinale exsudaten, droge ogen, sclerale geelzucht, retinale bloeding
Oor- en labyrintaandoeningen
gemeenschappelijk
Duizeligheid
Cardiale pathologieën
gemeenschappelijk
Hartkloppingen
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen
Heel gewoon
Hoest
gemeenschappelijk
Dyspneu, orofaryngeale pijn, dyspneu bij inspanning, productieve hoest
Maagdarmstelselaandoeningen
Heel gewoon
Misselijkheid, diarree
gemeenschappelijk
Braken, ascites, buikpijn, pijn in de bovenbuik, dyspepsie, droge mond, constipatie, opgezette buik, kiespijn, stomatitis, oesofageale refluxziekte, aambeien, buikpijn, gastritis, slokdarmvarices, afteuze stomatitis, slokdarmvarices
Lever- en galaandoeningen
gemeenschappelijk
Hyperbilirubinemie, geelzucht, poortadertrombose, leverfalen, door geneesmiddelen veroorzaakt leverletsel
Huid- en onderhuidaandoeningen
Heel gewoon
Jeuk, alopecia
gemeenschappelijk
Huiduitslag, droge huid, eczeem, jeukende uitslag, erytheem, hyperhidrose, algemene jeuk, nachtelijk zweten, huidlaesies
Ongewoon
Huidverkleuring, hyperpigmentatie van de huid
Skeletspierstelsel- en bindweefselaandoeningen
Heel gewoon
Spierpijn
gemeenschappelijk
Artralgie, spierspasmen, rugpijn, pijn in extremiteit, musculoskeletale pijn, botpijn
Nier- en urinewegaandoeningen
Ongewoon
Dysurie
Algemene aandoeningen en toedieningsplaatsstoornissen
Heel gewoon
Pyrexie, vermoeidheid, griepachtige ziekte, asthenie, koude rillingen, perifeer oedeem
gemeenschappelijk
Prikkelbaarheid, pijn, malaise, reacties op de injectieplaats, niet-cardiale pijn op de borst, oedeem, uitslag op de injectieplaats, ongemak op de borst, pruritus op de injectieplaats
Diagnostische toetsen
gemeenschappelijk
Bloedbilirubine verhoogd, gewicht verlaagd, aantal witte bloedcellen verlaagd, hemoglobine verlaagd, aantal neutrofielen verlaagd, international normalized ratio (INR) verhoogd, geactiveerde partiële tromboplastinetijd verlengd, verhoogde bloedglucose, bloedalbumine verlaging, QT-verlenging in het elektrocardiogram
Populatie van de klinische studie in AAS
Aandoeningen van het bloed en het lymfestelsel
gemeenschappelijk
Neutropenie, miltinfarct
Metabolisme en voedingsstoornissen
gemeenschappelijk
IJzerstapeling, verlies van eetlust, hypoglykemie, verhoogde eetlust
Psychische stoornissen
Heel gewoon
Slapeloosheid
gemeenschappelijk
Angst, depressie
Zenuwstelselaandoeningen
Heel gewoon
Hoofdpijn, duizeligheid
gemeenschappelijk
Syncope
Oogaandoeningen
gemeenschappelijk
Droge ogen, jeukende ogen, cataract, ooggeelzucht, wazig zien, slechtziendheid, floaters
Ademhalingsstelsel-, borstkas- en mediastinumaandoeningen
Heel gewoon
Hoesten, dyspneu, orofarynxpijn, rinorroe
gemeenschappelijk
Epistaxis
Maagdarmstelselaandoeningen
Heel gewoon
Buikpijn, diarree, misselijkheid
gemeenschappelijk
Bloedend tandvlees, blaren op het mondslijmvlies, orale pijn, braken, buikpijn, buikpijn, constipatie, opgezette buik, dysfagie, verkleurde ontlasting, gezwollen tong, stoornissen in de darmmotiliteit, winderigheid
Lever- en galaandoeningen
Heel gewoon
Verhoogde transaminasen
gemeenschappelijk
Verhoogd bloedbilirubine (hyperbilirubinemie), geelzucht
Niet bekend
Geneesmiddelgeïnduceerde leverbeschadiging *
* Gevallen van geneesmiddelgeïnduceerde leverbeschadiging zijn gemeld bij ITP- en HCV-patiënten
Huid- en onderhuidaandoeningen
Heel gewoon
Kneuzingen
gemeenschappelijk
Petechiën, uitslag, jeuk, netelroos, huidlaesies, maculaire uitslag
Ongewoon
Huidverkleuring, hyperpigmentatie van de huid
Skeletspierstelsel- en bindweefselaandoeningen
Heel gewoon
Artralgie, spierspasmen, pijn in extremiteiten
gemeenschappelijk
Rugpijn, spierpijn, botpijn
Nier- en urinewegaandoeningen
gemeenschappelijk
Chromaturie
Algemene aandoeningen en toedieningsplaatsstoornissen
Heel gewoon
Vermoeidheid, febriele neutropenie, koorts
gemeenschappelijk
Asthenie, perifeer oedeem, koude rillingen, malaise
Diagnostische toetsen
gemeenschappelijk
Verhoogd creatininefosfokinase in bloed
Beschrijving van geselecteerde bijwerkingen
Trombotische/trombo-embolische voorvallen (TEE)
In 3 gecontroleerde en 2 ongecontroleerde klinische onderzoeken bij volwassen patiënten met chronische ITP die eltrombopag kregen (n = 446), ondervonden 17 proefpersonen in totaal 19 trombo-embolische voorvallen, waaronder (in volgorde van afnemende frequentie) diepe veneuze trombose (n = 6) , longembolie (n = 6), acuut myocardinfarct (n = 2), herseninfarct (n = 2), embolie (n = 1) (zie rubriek 4.4).
In een placebogecontroleerd onderzoek (n = 288, veiligheidspopulatie) ondervonden 6 van de 143 (4%) volwassen patiënten met chronische leverziekte die eltrombopag kregen na 2 weken behandeling ter voorbereiding op invasieve procedures 7 TEE's. van 145 proefpersonen (1%) in de placebogroep had 3 TEE's. Vijf van de 6 met eltrombopag behandelde patiënten hadden TEE's met een trombocytenaantal > 200.000/l.
Er werden geen specifieke risicofactoren vastgesteld bij de proefpersonen die een TEE doormaakten, behalve een aantal bloedplaatjes ≥ 200.000 / l (zie rubriek 4.4).
In gecontroleerde onderzoeken bij trombocytopenische HCV-geïnfecteerde patiënten (n = 1439) hadden 38 van de 955 (4%) proefpersonen die werden behandeld met eltrombopag TEE's en 6 van de 484 proefpersonen (1%) in de placebogroep hadden TEE's. Poortadertrombose was de meest voorkomende TEE in beide behandelingsgroepen (2% bij patiënten behandeld met eltrombopag versus
Leverfalen (gebruik met interferon)
Chronische HCV-hepatitispatiënten met cirrose kunnen een risico lopen op leverdecompensatie wanneer zij alfa-interferontherapie krijgen. In 2 gecontroleerde klinische onderzoeken bij trombocytopenische patiënten met HCV-infectie werd leverdecompensatie (ascites, hepatische encefalopathie, varicesbloeding, spontane bacteriële peritonitis) vaker gemeld in de eltrombopag-arm (11%) dan in de placebo-arm (6%). Bij patiënten met lage albuminespiegels (≤ 35 g/l) of MELD-score ≥ 10 bij baseline, was er een driemaal hoger risico op leverdecompensatie en een verhoogd risico op fatale bijwerkingen in vergelijking met patiënten met minder leverziekte. Eltrombopag mag alleen aan dergelijke patiënten worden toegediend na zorgvuldige afweging van de verwachte voordelen versus de risico's. Patiënten met deze kenmerken moeten zorgvuldig worden gecontroleerd op tekenen en symptomen van leverdecompensatie (zie rubriek 4.4).
Trombocytopenie na stopzetting van de behandeling
In de 3 ITP-gecontroleerde klinische onderzoeken werden voorbijgaande dalingen van het aantal bloedplaatjes tot onder de uitgangswaarden waargenomen na stopzetting van de behandeling bij respectievelijk 8% van de eltrombopag-groep en 8% van de placebogroep (zie rubriek 4.4).
Verhoogd reticuline in het beenmerg
Binnen het programma waren bij geen van de patiënten klinisch relevante beenmergafwijkingen of klinische tekenen die wijzen op beenmergdisfunctie Bij een klein aantal patiënten met ITP werd de behandeling met eltrombopag stopgezet vanwege beenmergreticuline (zie rubriek 4.4).
Cytogenetische afwijkingen
In de eenarmige, open-label klinische studie bij AAS ondergingen patiënten beenmergaspiraat voor evaluatie van cytogenetische afwijkingen. Een nieuwe cytogenetische afwijking werd gemeld bij acht (19%) patiënten, waaronder 5 patiënten bij wie een wijziging van chromosoom 7. In de twee lopende onderzoeken (ELT116826 en ELT116643) werden cytogenetische afwijkingen gevonden bij 4/28 (14%) en respectievelijk 4/62 (6%) proefpersonen.
Hematologische neoplasmata
Bij drie (7%) patiënten in het eenarmige, open-label klinische AAS-onderzoek werd MDS gediagnosticeerd na behandeling met eltrombopag, in de twee lopende onderzoeken (ELT116826 en ELT116643) werd MDS of AML gediagnosticeerd bij 1 / 28 (4 %) en 1/62 (2%) proefpersonen in elk onderzoek.
Melding van vermoedelijke bijwerkingen
Het melden van vermoedelijke bijwerkingen die optreden na toelating van het geneesmiddel is belangrijk, omdat het een continue controle van de baten/risicoverhouding van het geneesmiddel mogelijk maakt.Beroepsbeoefenaren in de gezondheidszorg wordt verzocht alle vermoedelijke bijwerkingen te melden via het Italiaanse Geneesmiddelenbureau. , website: www.agenziafarmaco.gov.it/it/responsabili.
04.9 Overdosering
In het geval van een overdosis kan het aantal bloedplaatjes excessief toenemen en leiden tot trombotische/trombo-embolische complicaties. In geval van overdosering dient orale toediening te worden overwogen van een preparaat dat een metaalkation bevat, zoals calcium-, aluminium- of magnesiumpreparaten, om eltrombopag te chelateren en zo de absorptie ervan te beperken. Het aantal bloedplaatjes moet nauwlettend worden gecontroleerd. dient opnieuw te worden gestart volgens de doserings- en toedieningsaanbevelingen (zie rubriek 4.2).
In klinische onderzoeken was er een melding van overdosering waarbij de proefpersoon 5000 mg eltrombopag had ingenomen. De gemelde bijwerkingen waren onder meer lichte huiduitslag, voorbijgaande bradycardie, verhoging van ALAT en ASAT en vermoeidheid. Leverenzymen gemeten tussen dag 2 en 18 na inname hadden een piek in AST bij 1,6 keer l "ULN en ALT bij 3,9 keer l" ULN, en totaal bilirubine bij 2,4 keer l. "ULN. Het aantal bloedplaatjes was 672.000 / l op dag 18 na inname en het maximale aantal bloedplaatjes was 929.000 / l. Alle voorvallen verdwenen zonder gevolgen na de behandeling.
Aangezien eltrombopag niet significant via de nieren wordt uitgescheiden en sterk gebonden is aan plasma-eiwitten, wordt niet verwacht dat hemodialyse een effectieve methode is om de eliminatie van eltrombopag te verhogen.
05.0 FARMACOLOGISCHE EIGENSCHAPPEN
05.1 Farmacodynamische eigenschappen
Farmacotherapeutische categorie: antihemorragica, andere systemische hemostatica.
ATC-code: B02BX 05.
Werkingsmechanisme
Trombopoëtine (TPO) is het belangrijkste cytokine dat betrokken is bij de regulatie van megakarypoiese en bij de productie van bloedplaatjes, en is de endogene ligant voor de TPO-receptor (TPO-R). Eltrombopag interageert met het transmembraandomein van humaan TPO-R en initieert de signaalcascade vergelijkbaar maar niet identiek aan die van endogeen TPO, waardoor proliferatie en differentiatie van beenmergvoorlopercellen wordt geïnduceerd.
Klinische werkzaamheid en veiligheid
Studies bij chronische auto-immuun (idiopathische) trombocytopenie (ITP)
Twee fase III, gerandomiseerde, dubbelblinde, placebogecontroleerde onderzoeken, RAISE (TRA102537) en TRA100773B en twee open-label onderzoeken, REPEAT (TRA108057) en EXTEND (TRA105325) evalueerden de veiligheid en werkzaamheid van eltrombopag bij volwassen patiënten met eerder behandelde chronische ITP.
In totaal werd eltrombopag toegediend aan 277 patiënten met ITP gedurende ten minste 6 maanden en aan 202 patiënten gedurende ten minste 1 jaar.
Dubbelblinde placebogecontroleerde onderzoeken
RAISE: 197 patiënten met ITP werden gerandomiseerd in een verhouding van 2: 1 naar eltrombopag (n = 135) en placebo (n = 62), en de randomisatie werd gestratificeerd naar splenectomie, baselinegebruik voor TPI en baseline bloedplaatjestelling. aangepast gedurende de behandelingsperiode van 6 maanden op basis van het individuele aantal bloedplaatjes Alle patiënten begonnen met 50 mg eltrombopag Dag 29 tot het einde van de behandeling, van dag 15 tot het einde van de behandeling 28% van de met eltrombopag behandelde patiënten werd gehandhaafd op ≤ 25 mg en 29 tot 53% kreeg 75 mg.
Bovendien konden patiënten geleidelijk afbouwen van gelijktijdige ITP-medicatie en reddingsbehandelingen krijgen zoals voorgeschreven door lokale behandelingsrichtlijnen. Meer dan de helft van alle patiënten in elke behandelingsgroep had ≥ 3 eerdere ITP-therapieën gehad en 36% had eerder splenectomie gehad.
Het mediane aantal bloedplaatjes bij aanvang was 16.000 / l voor beide behandelingsgroepen en in de eltrombopag-groep was het meer dan 50.000 / l bij alle bezoeken tijdens de therapie, beginnend op dag 15; het mediane aantal bloedplaatjes in de placebogroep bleef echter
Een respons op het aantal bloedplaatjes tussen 50.000-400.000 / l in afwezigheid van salvage-behandeling werd bereikt door significant hogere aantallen patiënten in de eltrombopag-groep tijdens de behandelingsperiode van 6 maanden, p
Tabel 4: Secundaire werkzaamheidsresultaten van het RAISE-onderzoek
een logistiek regressiemodel gecorrigeerd voor gerandomiseerde stratificatievariabelen
b 21 van de 63 (33%) van de met eltrombopag behandelde patiënten die een basis- ITP-geneesmiddel gebruikten, stopten definitief met alle basis- ITP-geneesmiddelen.
Bij aanvang meldde meer dan 70% van de ITP-patiënten in elke behandelingsgroep bloedingen van elk type (WHO-graad 1-4) en meer dan 20% rapporteerde respectievelijk klinisch significante bloedingen (WHO-graad 2-4). Het percentage met eltrombopag behandelde patiënten met bloedingen van elk type (graad 1-4) en klinisch significante bloedingen (graad 2-4) daalde vanaf de uitgangswaarde met ongeveer 50% vanaf dag 15 tot het einde van de behandeling gedurende de behandelingsperiode van 6 maanden. .
TRA100773B: L "eindpunt primaire werkzaamheid was het aandeel van reageerders, gedefinieerd als patiënten met ITP met een toename van het aantal bloedplaatjes tot ≥ 50.000 / l op dag 43 vanaf een uitgangswaarde van 200.000 / l reageerders, werden degenen die om een andere reden stopten in aanmerking genomen non-responders ongeacht het aantal bloedplaatjes. Een totaal van 114 patiënten met eerder behandelde chronische ITP werden 2:1 gerandomiseerd naar eltrombopag (n = 76) en placebo (n = 38).
Tabel 5: Werkzaamheidsresultaten van TRA100773B-onderzoek
a - Logistiek regressiemodel gecorrigeerd voor gerandomiseerde stratificatievariabelen
In zowel RAISE als TRA100773B was de respons op eltrombopag vergeleken met placebo vergelijkbaar, ongeacht het gebruikte ITP-medicijn, splenectomie en het aantal bloedplaatjes op baseline (≤ 15.000 / μl,> 15.000 / l) bij randomisatie.
In de onderzoeken RAISE en TRA100773B in de subgroep van ITP-patiënten met een baseline trombocytenaantal ≤ 15.000 / l werd het vereiste niveau (> 50.000 / l) van het mediane aantal trombocyten niet bereikt, hoewel in beide onderzoeken 43% van deze met eltrombopag behandelde patiënten reageerde aan het einde van de behandelingsperiode van 6 weken. Bovendien reageerde in de RAISE-studie 42% van de patiënten met een baseline trombocytenaantal ≤ 15.000/μl behandeld met eltrombopag aan het einde van de behandelingsperiode van 6 maanden. Tweeënveertig tot 60% van de met eltrombopag behandelde patiënten in het RAISE-onderzoek ontving 75 mg vanaf dag 29 tot het einde van de behandeling.
Een open-label onderzoek met herhaalde dosering (3 kuren van 6 weken behandeling, afgewisseld met 4 weken zonder behandeling) toonde aan dat episodisch gebruik met meerdere kuren eltrombopag niet resulteerde in een vermindering van de respons.
Eltrombopag werd toegediend aan 302 patiënten met ITP in de open-label vervolgstudie EXTEND (TRA105325), 218 voltooiden 1 jaar, 180 voltooiden 2 jaar, 107 voltooiden 3 jaar, 75 voltooiden 4 jaar, 34 voltooiden 5 jaar en 18 voltooiden 6 jaar. Het mediane aantal bloedplaatjes bij baseline was 19.000/μl voorafgaand aan de toediening van eltrombopag. Het mediane aantal bloedplaatjes na 1, 2, 3, 4, 5, 6 en 7 jaar van het onderzoek was 85.000 / microliter, 85.000 / microliter, 105.000 / microliter, 64.000 / microliter, 75.000 / microliter, 119.000 / microliter en 76.000 / microliter microliter, respectievelijk.
Er zijn geen klinische onderzoeken uitgevoerd waarin eltrombopag werd vergeleken met andere therapeutische opties (bijv. splenectomie). De veiligheid van eltrombopag op de lange termijn moet worden overwogen voordat met de behandeling wordt begonnen.
Pediatrische patiënten (leeftijd 1-17 jaar)
De veiligheid en werkzaamheid van eltrombopag bij pediatrische proefpersonen werd geëvalueerd in twee onderzoeken. TRA115450 (PETIT2): Het primaire eindpunt was aanhoudende respons, gedefinieerd als het aandeel van de met eltrombopag behandelde proefpersonen in vergelijking met placebo, die gedurende ten minste 6 van de 8 weken (bij afwezigheid van salvage-therapie) een bloedplaatjesaantal van ≥ 50.000 / mcl bereikten, in de weken 5 en 12 tijdens de gerandomiseerde dubbelblinde periode Proefpersonen bij wie gedurende ten minste 1 jaar chronische ITP was gediagnosticeerd en die ongevoelig waren voor of recidiveerden bij ten minste één eerdere ITP-therapie of die om een bepaalde reden andere ITP-behandelingen niet konden voortzetten en een aantal bloedplaatjes hadden
Over het algemeen bereikte een significant hoger percentage proefpersonen in de eltrombopag-groep (40%) het primaire eindpunt (odds ratio: 18,0 [95% BI: 2,3; 140,9] p
Tabel 6: Aanhoudende bloedplaatjesresponspercentages per cohortleeftijd bij pediatrische proefpersonen met chronische ITP
Statistisch gezien hadden minder met eltrombopag behandelde proefpersonen noodbehandeling nodig tijdens de gerandomiseerde periode dan met placebo behandelde proefpersonen (19% [12/63] vs 24% [7/29], p = 0,032).
Bij aanvang meldde 71% van de proefpersonen in de eltrombopag-groep en 69% in de placebogroep bloedingen (WHO-graden 1-4). In week 12 was het aantal met eltrombopag behandelde proefpersonen dat bloeding meldde gehalveerd ten opzichte van de uitgangswaarde (36%). Ter vergelijking: in week 12 meldde 55% van de met placebo behandelde proefpersonen geen bloeding.
De proefpersonen mochten de achtergrondtherapie voor ITP alleen verminderen of stopzetten tijdens de open-labelfase van het onderzoek, en 53% (8/15) van de proefpersonen kon de basistherapie verminderen (n = 1) of stopzetten (n = 7) voor ITP, voornamelijk corticosteroïden, zonder de noodzaak van noodtherapie.
TRA108062 (PETIT): Het primaire eindpunt was het percentage proefpersonen dat ten minste eenmaal een bloedplaatjesaantal van ≥ 50.000 / mcl bereikte tussen week 1 en 6. De proefpersonen waren ongevoelig voor of recidiveerden bij ten minste één eerdere ITP-therapie en met een bloedplaatjes
Over het algemeen bereikte een significant hoger percentage proefpersonen in de eltrombopag-groep (62%) het primaire eindpunt (Odds Ratio: 4,3 [95% BI: 1,4, 13,3] p = 0,011).
Aanhoudende respons werd waargenomen bij 50% van de eerste respondenten in 20 van 24 weken in de PETIT 2-studie en 15 van 24 weken in de PETIT-studie.
Onderzoek naar trombocytopenie geassocieerd met chronische HCV-hepatitis
De werkzaamheid en veiligheid van eltrombopag voor de behandeling van trombocytopenie bij met HCV geïnfecteerde patiënten werden geëvalueerd in twee gerandomiseerde, dubbelblinde, placebogecontroleerde onderzoeken.ENABLE 1 gebruikte peginterferon alfa-2a plus ribavirine voor antivirale behandeling en ENABLE 2 gebruikte peginterferon alfa- 2b plus ribavirine Patiënten kregen geen direct werkende antivirale middelen In beide onderzoeken werden patiënten met gescreend aantal bloedplaatjes geïncludeerd (
Ziektekenmerken bij baseline waren vergelijkbaar in beide onderzoeken en kwamen overeen met de met HCV geïnfecteerde patiëntenpopulatie met gecompenseerde cirrose. De meeste patiënten hadden HCV-genotype 1 (64%) en hadden overbruggende fibrose/cirrose. Eenendertig procent van de patiënten was eerder behandeld met therapieën voor HCV-infectie, voornamelijk gepegyleerd interferon plus ribavirine Het mediane aantal bloedplaatjes bij baseline was 59.500 / l in beide behandelingsgroepen: 0,8%, 28% en 72% van de gerekruteerde patiënten had bloedplaatjes telt
De onderzoeken bestonden uit twee fasen: een antivirale voorbehandelingsfase en een antivirale behandelingsfase.In de antivirale voorbehandelingsfase kregen proefpersonen open-label eltrombopag om het aantal bloedplaatjes te verhogen tot ≥ 90.000 / mcl voor ENABLE 1 en ≥ 100.000 / mcl voor ENABLE 2. Mediane tijd om het doel van het aantal bloedplaatjes van ≥ 90.000 / mcl te bereiken (ENABLE 1 ) of ≥ 100.000 / mcl (ENABLE 2) was 2 weken.
L"eindpunt primaire werkzaamheid voor beide onderzoeken was aanhoudende virologische respons (gestaag virologische reactie, SVR), gedefinieerd als het percentage patiënten met een HCV-infectie met niet-detecteerbaar HCV-RNA 24 weken na voltooiing van de geplande behandelingsperiode.
In beide met HCV geïnfecteerde onderzoeken bereikte een significant hoger percentage van de met eltrombopag behandelde patiënten (n = 201, 21%) SVR vergeleken met degenen die werden behandeld met placebo (n = 65, 13%) (zie tabel 7). De verbetering in het percentage patiënten dat SVR bereikte, was consistent in alle subgroepen in de randomisatiestrata (bloedplaatjes bij baseline (vs.> 50.000), viral load (vs. ≥ 800.000 IE/ml) en genotype (2/3 vs. 1/4/6).
Tabel 7: Virologische respons van patiënten met HCV-infectie in ENABLE 1 en ENABLE 2
a Eltrombopag gegeven in combinatie met peginterferon alfa-2a (180 g eenmaal per week gedurende 48 weken voor genotypen 1/4/6; gedurende 24 weken voor genotypen 2/3) plus ribavirine (vanaf 800
bij 1200 mg per dag in 2 verdeelde doses oraal)
b Eltrombopag toegediend in combinatie met peginterferon-alfa-2b (1,5 mcg/kg eenmaal per week gedurende 48 weken voor genotype 1/4/6; gedurende 24 weken voor genotype 2/3) plus ribavirine (800 tot 1400 mg oraal in 2 verdeelde doses)
c Het beoogde aantal bloedplaatjes was ≥ 90.000 / mcl voor ENABLE 1 en ≥ 100.000 / mcl voor ENABLE 2. Voor ENABLE werden 1 682 patiënten gerandomiseerd naar de antivirale behandelingsfase; 2 proefpersonen trokken echter hun toestemming in voordat ze antivirale therapie kregen.
d waarde P versus placebo
en 64% van de proefpersonen die deelnamen aan de ENABLE 1- en ENABLE 2-studies had genotype 1
F post-hoc analyseert
Andere secundaire observaties uit de onderzoeken waren de volgende: Aanzienlijk minder patiënten die werden behandeld met eltrombopag hadden de antivirale therapie voortijdig stopgezet in vergelijking met degenen die werden behandeld met placebo (45% vs. 60%, p = versus 27%). Behandeling met eltrombopag vertraagde en verminderde het aantal dosisverlagingen van peginterferon.
Ernstige aplastische anemie
Eltrombopag werd onderzocht in een eenarmige, single-center, open-label klinische studie bij 43 patiënten met ernstige aplastische anemie met refractaire trombocytopenie na ten minste één eerdere immunosuppressieve therapie (soa) en met een aantal bloedplaatjes ≤ 30.000/μl.
De meeste proefpersonen, 33 (77%), werden beschouwd als "primaire refractaire ziekte", gedefinieerd als de "afwezigheid van een adequate eerste respons op immunosuppressieve therapie in welke lijn dan ook. De overige 10 proefpersonen hadden een bloedplaatjesrespons. onvoldoende op eerdere Alle 10 die werden behandeld, hadden ten minste 2 eerdere immunosuppressieve therapieregimes ondergaan en 50% had ten minste 3 eerdere immunosuppressieve therapieregimes gekregen. Patiënten bij wie anemie werd vastgesteld
Fanconi, een infectie die niet reageerde op geschikte therapie, een PNH-kloon in neutrofielen met een grootte van ≥ 50%, werd uitgesloten van het onderzoek.
Bij baseline was het mediane aantal bloedplaatjes 20.000 / μl, het hemoglobine was 8,4 g / dl, het absolute aantal neutrofielen was 0,58 x 109 / l en het absolute aantal reticulocyten was 24,3 x 109 / 1. Zesentachtig procent van de patiënten was afhankelijk van erytrocyten transfusies en 91% was afhankelijk van trombocytentransfusies De meeste patiënten (84%) hadden ten minste 2 eerdere immunosuppressieve therapieën ontvangen Drie patiënten hadden bij aanvang cytogenetische afwijkingen.
Het primaire eindpunt was hematologische respons beoordeeld na 12 weken behandeling met eltrombopag.Hematologische respons werd gedefinieerd als het bereiken van een of meer van de volgende criteria: 1) verhoging van het aantal bloedplaatjes tot 20.000 / l boven de uitgangswaarde of het aantal bloedplaatjes stabiel met transfusie-onafhankelijkheid voor minimaal 8 weken 2) een verhoging van het hemoglobinegehalte van > 1,5 g/dl, of een verlaging van de transfusies van ≥ 4 eenheden rode bloedcellen (RBC) gedurende 8 opeenvolgende weken; 3) toename van het absolute aantal neutrofielen (ANC) met 100% of toename van ANC> 0,5 x 109 / L.
Het hematologische responspercentage was 40% (17/43 patiënten; 95% BI 25, 56), en de meeste responsen waren beperkt tot een enkele lijn (13 / 17,76%), terwijl er 3 responsen werden geregistreerd. -lineaire respons in week 12. Eltrombopag werd na 16 weken stopgezet als er geen hematologische respons of transfusie-onafhankelijkheid werd waargenomen. Responders zetten de therapie voort in een verlengingsfase van het onderzoek. In totaal namen 14 patiënten deel aan de verlengingsfase van het onderzoek. Negen van deze patiënten bereikten een multilineaire respons, 4 van de 9 zetten de behandeling voort en 5 verlaagden de behandeling met eltrombopag en behielden de respons (mediaan follow-up: 20,6 maanden, bereik: 5,7 tot 22,5 maanden) De overige 5 patiënten stopten met de behandeling, drie vanwege een terugval waargenomen bij het bezoek van de derde maand van de verlengingsfase.
Tijdens de behandeling met eltrombopag werd 59% (23/39) onafhankelijk van trombocytentransfusie (28 dagen zonder trombocytentransfusie) en 27% (10/37) onafhankelijk van RBC-transfusie (56 dagen zonder transfusie van RBC). De langste trombocytentransfusievrije periode voor non-responders was 27 dagen (mediaan). De langste trombocytentransfusievrije periode voor responders was 29 dagen (mediaan). De langste erytrocytentransfusievrije periode voor responders was 266 dagen (mediaan).
Meer dan 50% van de responders die bij baseline transfusieafhankelijk waren, had een afname van >80% in de behoefte aan zowel bloedplaatjes- als RBC-transfusies vanaf baseline.
Voorlopige resultaten van een ondersteunend onderzoek (onderzoek ELT116826), een lopend niet-gerandomiseerd, fase II, eenarmig, open-label onderzoek bij refractaire proefpersonen met AAS, lieten consistente resultaten zien. De gegevens zijn beperkt tot 21 van de 60 voorspelde patiënten en een hematologische respons werd gezien bij 52% van de patiënten na 6 maanden. Bij 45% van de patiënten werden multilineaire responsen gemeld.
05.2 Farmacokinetische eigenschappen
Farmacokinetiek
Eltrombopag-plasmaconcentratiegegevens - tijd verzameld bij 88 ITP-patiënten in TRA100773A en TRA100773B werden gecombineerd met gegevens van 111 gezonde volwassen proefpersonen in een farmacokinetische populatieanalyse. Schattingen van de plasma-AUC (0-?) En Cmax-waarden van eltrombopag bij patiënten met ITP worden weergegeven (tabel 8).
Tabel 8: Geometrisch gemiddelde (95% betrouwbaarheidsinterval) van farmacokinetische parameters in plasma van eltrombopag bij steady-state bij volwassenen met ITP
a - Schattingen van de AUC (0-?) en Cmax op basis van post-hoc farmacokinetische populatiewaarden.
Gegevens over plasmaconcentraties van eltrombopag in de loop van de tijd verzameld bij 590 met HCV geïnfecteerde proefpersonen die deelnamen aan de fase III-onderzoeken TPL103922 / ENABLE 1 en TPL108390 / ENABLE 2 werden gecombineerd met gegevens van met HCV geïnfecteerde patiënten die deelnamen aan de fase II-studie TPL102357 en gezonde volwassen proefpersonen in een farmacokinetische populatieanalyse Schattingen van de Cmax en AUC (0-?) van eltrombopag plasma bij met HCV geïnfecteerde patiënten die deelnamen aan fase 3-onderzoeken worden voor elke dosis vermeld in tabel 9.
Tabel 9 Geometrisch gemiddelde (95% BI) allo stabiele toestand eltrombopag plasma farmacokinetische parameters bij patiënten met chronische HCV-infectie
Gegevens gepresenteerd als geometrisch gemiddelde (95% BI).
AUC (0-?) En Cmax op basis van schattingen post-hoc populatiefarmacokinetiek bij de hoogste dosis in elke patiëntgegevens.
Absorptie en biologische beschikbaarheid
Eltrombopag wordt geabsorbeerd met een piekconcentratie die 2 tot 6 uur na orale toediening optreedt. Toediening van eltrombopag gelijktijdig met antacida en andere producten die polyvalente kationen bevatten, zoals zuivelproducten en mineraalsupplementen, vermindert de blootstelling aan eltrombopag aanzienlijk (zie rubriek 4.2).. In een relatieve biologische beschikbaarheidsstudie bij volwassenen bereikte eltrombopag poeder voor orale suspensie een 22% hogere plasma-AUC (0-?) in vergelijking met de tabletformulering. De absolute orale biologische beschikbaarheid van eltrombopag na toediening bij de mens is niet vastgesteld. 52%.
Verdeling
Eltrombopag is sterk gebonden aan humane plasma-eiwitten (> 99,9%), voornamelijk albumine Eltrombopag is een BCRP-substraat, maar is geen P-glycoproteïne of OATP1B1-substraat.
Biotransformatie
Eltrombopag wordt voornamelijk gemetaboliseerd door splitsing, oxidatie en conjugatie met glucuronzuur, glutathion of cysteïne. In een radioactief gelabeld onderzoek bij mensen is eltrombopag goed voor ongeveer 64% van de AUC0-? plasmaconcentratie van radioactief gemerkte steenkool. Kleine metabolieten als gevolg van glucuronidering en oxidatie werden ook gevonden. Opleiding in vitro suggereren dat CYP1A2 en CYP2C8 verantwoordelijk zijn voor het oxidatieve metabolisme van eltrombopag. De uridinedifosfoglucuronyltransferasen UGT1A1 en UGT1A3 zijn verantwoordelijk voor glucuronidering, en bacteriën uit het lagere maagdarmkanaal kunnen verantwoordelijk zijn voor splitsing.
Eliminatie
Eenmaal geabsorbeerd, wordt eltrombopag uitgebreid gemetaboliseerd. De belangrijkste uitscheidingsroute van eltrombopag is via de feces (59%) waarbij 31% van de dosis als metabolieten in de urine wordt aangetroffen. De onveranderde verbinding (eltrombopag) wordt niet in de urine aangetroffen. Onveranderd eltrombopag dat in de feces wordt uitgescheiden, vertegenwoordigt ongeveer 20% van de dosis. De plasma-eliminatiehalfwaardetijd van eltrombopag is ongeveer 21-32 uur.
Farmacokinetische interacties
Gebaseerd op een onderzoek bij mensen met radioactief gelabeld eltrombopag, speelt glucuronidering een ondergeschikte rol in het metabolisme van eltrombopag. Studies in humane levermicrosoom hebben UGT1A1 en UGT1A3 geïdentificeerd als de enzymen die verantwoordelijk zijn voor de glucuronidering van eltrombopag. Eltrombopag is een remmer van verschillende UGT-enzymen. in vitro. Klinisch significante geneesmiddelinteracties waarbij glucuronidering betrokken is, worden niet verwacht vanwege de beperkte bijdrage van individuele UGT-enzymen aan eltrombopag-glucuronidering en mogelijke gelijktijdig toegediende geneesmiddelen.
Ongeveer 21% van een dosis eltrombopag kan oxidatief worden gemetaboliseerd. Studies in menselijke levermicrosoom hebben CYP1A2 en CYP2C8 geïdentificeerd als de enzymen die verantwoordelijk zijn voor de oxidatie van eltrombopag. Op basis van gegevens remt of induceert eltrombopag geen CYP-enzymen in vitro en in vivo (zie rubriek 4.5)
Opleiding in vitro heeft aangetoond dat eltrombopag een remmer is van de transporter OATP1B1 en een remmer van de transporter BCRP en eltrombopag in een klinisch interactieonderzoek de blootstelling aan OATP1B1 en BCRP van het rosuvastatinesubstraat verhoogt (zie rubriek 4.5). was 50% dosisverlaging van statines aanbevolen Gelijktijdige toediening van 200 mg ciclosporine (BCRP-remmer) verlaagde de Cmax en AUCinf van eltrombopag met respectievelijk 25% en 18% Gelijktijdige toediening van 600 mg ciclosporine verlaagde de Cmax en AUCinf van eltrombopag met 39% en 24%, respectievelijk.
Eltrombopag cheleert polyvalente kationen zoals ijzer, calcium, magnesium, aluminium, selenium en zink (zie rubrieken 4.2 en 4.5).
Toediening van een enkelvoudige dosis van 50 mg eltrombopag in tabletten met een standaard calorierijke maaltijd, een vetrijk ontbijt met zuivelproducten, verminderde de gemiddelde plasma-AUC van eltrombopag met 59% en de Cmax met gemiddeld 65%.
Toediening van een enkelvoudige dosis van 25 mg eltrombopag poeder voor orale suspensie met een maaltijd met veel calcium, matig vet en calorieën verminderde de gemiddelde plasma-AUC0-? van eltrombopag met 75% en de gemiddelde Cmax met 79%. een enkele dosis van 25 mg eltrombopag poeder voor orale suspensie werd 2 uur vóór een calciumrijke maaltijd toegediend (gemiddelde AUC0-? verlaagd met 20% en gemiddelde Cmax van 14%).
Calciumarm voedsel (fruit, magere ham, rundvlees en vruchtensap zonder toegevoegd calcium, magnesium of ijzer), sojamelk zonder toegevoegd en tarwe heeft geen significante invloed op de plasmablootstelling van eltrombopag, ongeacht het calorie- en vetgehalte (zie rubrieken 4.2 en 4.5).
Speciale patiëntenpopulaties
Nierfalen
De farmacokinetiek van eltrombopag is onderzocht na toediening van eltrombopag aan volwassen proefpersonen met nierinsufficiëntie. Na toediening van een enkelvoudige dosis van 50 mg was de AUC0-? van eltrombopag 32% tot 36% lager bij proefpersonen met lichte tot matige nierinsufficiëntie en 60% lager bij proefpersonen met ernstige nierinsufficiëntie in vergelijking met gezonde vrijwilligers. variabiliteit en significante overlap in blootstellingen tussen patiënten met nierinsufficiëntie en gezonde vrijwilligers Vrije (actieve) eltrombopag-concentraties voor dit sterk eiwitgebonden geneesmiddel werden niet gemeten Patiënten met een verminderde nierfunctie dienen eltrombopag met voorzichtigheid te gebruiken en onder zorgvuldige controle, bijvoorbeeld door uitvoeren van serumcreatinine en/of urineonderzoek (zie rubriek 4.2) De werkzaamheid en veiligheid van eltrombopag zijn niet vastgesteld bij proefpersonen met zowel matige tot ernstige nierinsufficiëntie als leverinsufficiëntie.
Leverinsufficiëntie
De farmacokinetiek van eltrombopag is onderzocht na toediening van eltrombopag aan volwassen proefpersonen met leverinsufficiëntie. Na toediening van een enkelvoudige dosis van 50 mg was de AUC0-? van eltrombopag 41% hoger bij proefpersonen met lichte leverinsufficiëntie en 80% tot 93% hoger bij proefpersonen met matige tot matige leverinsufficiëntie ernstig, in vergelijking met gezonde vrijwilligers. Er was een aanzienlijke variabiliteit en een significante overlap in blootstellingen tussen patiënten met leverinsufficiëntie en gezonde vrijwilligers. Vrije (actieve) eltrombopag-concentraties voor dit sterk eiwitgebonden geneesmiddel zijn niet gemeten.
De invloed van leverinsufficiëntie op de farmacokinetiek van eltrombopag na herhaalde toediening werd geëvalueerd met behulp van een farmacokinetische populatieanalyse bij 28 gezonde volwassenen en 714 patiënten met leverinsufficiëntie (673 patiënten met HCV-infectie en 41 patiënten met chronische leverziekte van een andere etiologie). Van deze 714 patiënten hadden 642 lichte leverinsufficiëntie, 67 hadden matige leverinsufficiëntie en 2 hadden ernstige leverinsufficiëntie. In vergelijking met gezonde vrijwilligers hadden patiënten met een lichte leverfunctiestoornis plasma-eltrombopag AUC (0-?) waarden hoger dan ongeveer 111% (95% BI: 45% tot 283%) en patiënten met matige leverfunctiestoornis hadden plasmawaarden eltrombopag AUC ( 0-?) groter dan ongeveer 183% (95% BI: 90% tot 459%).
Daarom mag eltrombopag niet worden gebruikt bij ITP-patiënten met een matige tot ernstige leverfunctiestoornis (Child-Pugh-score ≥ 5), tenzij het verwachte voordeel opweegt tegen het vastgestelde risico op poortadertrombose (zie rubrieken 4.2 en 4.4). Voor met HCV geïnfecteerde patiënten start eltrombopag met een dosis van 25 mg eenmaal daags (zie rubriek 4.2).
Ras
De invloed van Oost-Aziatische etniciteit op de farmacokinetiek van eltrombopag werd beoordeeld met behulp van farmacokinetische populatieanalyse bij 111 gezonde volwassenen (31 Oost-Aziaten) en 88 patiënten met ITP (18 Oost-Aziaten). zoals Japanners, Chinezen, Taiwanezen en Koreanen) hadden ongeveer 49% hogere plasma-eltrombopag AUC (0-?) waarden vergeleken met niet-Oost-Aziatische patiënten, die overwegend blank waren (zie rubriek 4.2).
De invloed van Oost-Aziatische etniciteit (zoals Chinees, Japans, Taiwanees, Koreaans en Thais) op de farmacokinetiek van eltrombopag werd geëvalueerd met behulp van een farmacokinetische populatieanalyse bij 635 met HCV geïnfecteerde patiënten (145 Oost-Aziaten en 69 Zuidoost-Aziaten) Gebaseerd op schattingen van de populatie farmacokinetische analyse hadden patiënten van Oost-Aziatische etniciteit ongeveer 55% hogere plasma-eltrombopag AUC (0-?) waarden in vergelijking met patiënten van andere rassen, waarvan ze overwegend blank waren (zie rubriek 4.2).
Seks
De invloed van geslacht op de farmacokinetiek van eltrombopag werd geëvalueerd met behulp van farmacokinetische populatieanalyse bij 111 gezonde volwassenen (14 vrouwen) bij 88 ITP-patiënten (57 vrouwen). Gebaseerd op schattingen van farmacokinetische populatieanalyses, hadden vrouwelijke ITP-patiënten ongeveer 23% hogere plasma-eltrombopag AUC (0-?) waarden in vergelijking met mannelijke patiënten, zonder lichamelijke aanpassingen voor gewichtsverschillen.
De invloed van geslacht op de farmacokinetiek van eltrombopag werd geëvalueerd met behulp van farmacokinetische populatieanalyse bij 635 met HCV geïnfecteerde patiënten (260 vrouwen). Op basis van modelschattingen hadden vrouwelijke patiënten met een HCV-infectie ongeveer 41% hogere plasma-eltrombopag AUC (0-?) waarden in vergelijking met mannelijke patiënten.
Leeftijd
De invloed van leeftijd op de farmacokinetiek van eltrombopag werd geëvalueerd met behulp van populatiefarmacokinetische analyse bij 28 gezonde proefpersonen, 673 patiënten met een HCV-infectie en 41 patiënten met chronische leverziekte van andere etiologie met een leeftijdsbereik van 19. leeftijd 74. Er zijn geen farmacokinetische gegevens beschikbaar over de gebruik van eltrombopag bij patiënten 75 jaar. Op basis van modelschattingen hadden oudere patiënten (≥ 65 jaar) ongeveer 41% hogere plasma-eltrombopag AUC (0-?) waarden in vergelijking met jongere patiënten (zie rubriek 4.2).
Pediatrische patiënten (leeftijd 1-17 jaar)
De farmacokinetiek van eltrombopag werd geëvalueerd bij 168 pediatrische patiënten met ITP in twee eenmaal daagse onderzoeken, TRA108062 / PETIT en TRA115450 / PETIT-2. De schijnbare plasmaklaring van eltrombopag na orale toediening (CL/F) nam toe met toenemend lichaamsgewicht.
De effecten van ras en geslacht op plasma-eltrombopag CL/F-schattingen waren consistent tussen pediatrische en volwassen patiënten. Oost-Aziatische pediatrische ITP-patiënten hadden ongeveer 43% hogere plasma-eltrombopag-AUC (0-?) In vergelijking met niet-Oost-Aziatische patiënten. Vrouwelijke pediatrische ITP-patiënten hadden een toename van ongeveer 25% in plasma-AUC. "Plasma-AUC (0-?) Van eltrombopag vergeleken met mannelijke patiënten.
De farmacokinetische parameters van eltrombopag bij pediatrische patiënten met ITP worden weergegeven in tabel 10.
Tabel 10. Geometrisch gemiddelde (95% BI) van de farmacokinetische parameters bij steady-state van de plasmaconcentratie van eltrombopag bij pediatrische proefpersonen met ITP (50 mg eenmaal daags doseringsschema)
Gegevens gepresenteerd als geometrisch gemiddelde (95% BI). AUC (0-?) En Cmax zijn gebaseerd op farmacokinetische schattingen van de post-hocpopulatie.
05.3 Gegevens uit het preklinisch veiligheidsonderzoek
Omdat eltrombopag de aanmaak van bloedplaatjes bij muizen, ratten of honden niet stimuleert vanwege de specificiteit van de unieke TPO-receptor, vormen de gegevens van deze dieren geen volledig model voor het evalueren van de mogelijke farmacologiegerelateerde bijwerkingen van eltrombopag bij de mens. reproductie- en carcinogeniteitsstudies.
Aan de behandeling gerelateerde cataracten werden gedetecteerd bij knaagdieren en waren dosis- en tijdsafhankelijk. Bij blootstellingen van meer dan 6 keer de humane klinische blootstelling bij volwassen ITP-patiënten bij een dosis van 75 mg/dag en bij blootstellingen die 3 keer de humane klinische blootstelling bij HCV-geïnfecteerde volwassen patiënten bij 100 mg/dag, werden blootstellingen gebaseerd op AUC, cataract waargenomen bij muizen na 6 weken en bij ratten na behandeling van 28 weken Bij blootstellingen groter dan of gelijk aan 4 maal de klinische blootstelling bij de mens bij ITP-patiënten in een dosis van 75 mg/dag en a Blootstellingen 2 maal de klinische blootstelling bij de mens bij met HCV geïnfecteerde patiënten bij een dosis van 100 mg / dag, blootstellingen op basis van AUC, cataract werd waargenomen bij muizen na 13 weken en bij ratten na 39 weken behandeling. Bij niet-verdraagbare doses bij juveniele ratten vóór het spenen die werden toegediend van dag 4 tot 32 (ongeveer 2 mensenjaren aan het einde van de doseringsperiode), werden oculaire opaciteiten (histologie niet uitgevoerd) waargenomen bij gelijke doses van 75 mg / dag. 9 keer de maximale klinische blootstelling bij de mens bij pediatrische ITP-patiënten, gebaseerd op de AUC. Er werd echter geen cataract waargenomen bij juveniele ratten die getolereerde doses kregen van 5 keer de klinische blootstelling bij mensen bij pediatrische ITP-patiënten, gebaseerd op de AUC. Er werd geen cataract waargenomen bij volwassen honden na 52 weken behandeling (2 keer de klinische blootstelling bij mensen bij volwassen of pediatrische patiënten met ITP in een dosis van 75 mg/dag en equivalent aan de klinische blootstelling bij mensen bij patiënten met een HCV-infectie in een dosis van 100 mg/dag, gebaseerd op de AUC).
In onderzoeken bij muizen en ratten met een duur van maximaal 14 dagen werd niertubulaire toxiciteit waargenomen bij blootstellingen die over het algemeen geassocieerd waren met morbiditeit en mortaliteit. Tubulaire toxiciteit werd ook waargenomen in een 2 jaar durende orale carcinogeniteitsstudie bij muizen bij doses van 25, 75 en 150 mg/kg/dag. De effecten waren minder ernstig bij lagere doses en werden gekenmerkt door een reeks regeneratieve modificaties. De blootstelling bij de laagste dosis was 1,2 of 0,8 keer de klinische blootstelling bij mensen op basis van de AUC bij volwassen of pediatrische ITP-patiënten bij 75 mg/dag en 0,6 keer L. klinische blootstelling bij mensen bij HCV-patiënten bij een dosis van 100 mg/dag, blootstellingen op basis van AUC Niereffecten werden niet waargenomen bij ratten na 28 weken of bij honden na 52 weken bij blootstellingen van 4 en 2 maal de klinische blootstelling bij mensen bij volwassen ITP-patiënten en 3 en 2 maal de klinische blootstelling bij mensen bij pediatrische patiënten met ITP in een dosis van 75 mg/dag en bij blootstellingen van 2 maal en equivalent aan klinische blootstelling bij mensen bij patiënten met HCV in een dosis van 100 mg/dag, blootstellingen gebaseerd op AUC.
Hepatocytdegeneratie en/of necrose, vaak vergezeld van verhoogde serumleverenzymen, werd waargenomen bij muizen, ratten en honden in doses die geassocieerd waren met morbiditeit en mortaliteit of die slecht werden verdragen. Er werden geen effecten op de lever waargenomen na chronische behandeling bij ratten (28 weken) en honden (52 weken) bij blootstellingen van 4 of 2 maal de klinische blootstelling bij de mens bij volwassen patiënten met ITP en bij blootstellingen van 3 of 2 maal de klinische blootstelling mens bij pediatrische ITP-patiënten bij een dosis van 75 mg/dag en 2 maal of equivalent aan klinische blootstelling bij mensen bij met HCV geïnfecteerde patiënten bij 100 mg/dag, blootstellingen gebaseerd op AUC.
In kortetermijnstudies, bij slecht verdragen doses bij ratten en honden (blootstelling groter dan 10 of 7 maal de klinische blootstelling bij de mens bij volwassen of pediatrische ITP-patiënten in een dosis van 75 mg/dag en blootstelling van meer dan 4 maal de klinische blootstelling bij met HCV geïnfecteerde patiënten aan een dosis van 100 mg/dag, blootstellingen op basis van AUC), verminderde aantallen reticulocyten en regeneratieve erytroïde hyperplasie van het beenmerg (alleen ratten) Opmerkelijke effecten op het aantal rode bloedcellen of reticulocyten na behandeling gedurende maximaal 28 weken bij ratten, 52 weken bij honden en 2 jaar bij muizen of ratten bij maximaal verdraagbare doses, wat overeenkomt met een 2- tot 4-voudige blootstelling humane klinische blootstelling bij volwassen of pediatrische ITP-patiënten in een dosis van 75 mg/dag en bij blootstellingen van minder dan 2 maal de klinische blootstelling bij mensen bij met HCV geïnfecteerde patiënten in een dosis van 100 mg/dag, ionen op basis van AUC.
Endosteale hyperostose werd waargenomen in een 28 weken durende toxiciteitsstudie bij ratten bij een ondraaglijke dosis van 60 mg/kg/dag (6 maal of 4 maal de humane klinische blootstelling bij volwassen of pediatrische ITP-patiënten bij een dosis van 75 mg/dag en 3 maal de klinische blootstelling bij de mens bij met HCV geïnfecteerde patiënten bij een dosis van 100 mg/dag, blootstellingen gebaseerd op de AUC) Er werden geen botveranderingen waargenomen bij muizen of ratten na levenslange blootstelling (2 jaar) bij 4 of 2 maal de klinische blootstelling bij de mens bij volwassen of pediatrische ITP-patiënten in een dosis van 75 mg/dag en bij 2 maal de klinische blootstelling bij mensen bij patiënten met HCV in een dosis van 100 mg/dag, blootstellingen gebaseerd op de AUC.
Eltrombopag was niet carcinogeen bij muizen bij doseringen tot 75 mg/kg/dag of bij ratten bij doseringen tot 40 mg/kg/dag (blootstellingen tot 4 of 2 maal de humane klinische blootstelling bij volwassen of pediatrische patiënten met ITP bij een dosis van 75 mg/dag en 2 maal de humane klinische blootstelling bij met HCV geïnfecteerde patiënten bij een dosis van 100 mg/dag, blootstellingen gebaseerd op AUC Eltrombopag was niet mutageen of clastogeen in een testmutatie op bacteriën of in twee tests in vivo bij de rat (micronucleus en ongeplande DNA-synthese, 10 keer of 8 keer de menselijke klinische blootstelling bij volwassen of pediatrische ITP-patiënten in een dosis van 75 mg / dag en 7 keer de menselijke klinische blootstelling bij patiënten met HCV-infectie bij een dosis van 100 mg / dag, blootstellingen op basis van Cmax). In de test in vitro op muislymfoom was eltrombopag marginaal positief (verhoogde mutaties). deze observaties in vitro En in vivo suggereren dat eltrombopag geen genotoxisch risico vormt voor de mens.
Eltrombopag heeft geen invloed op de vrouwelijke vruchtbaarheid, vroege embryo-ontwikkeling of embryo-foetale ontwikkeling bij ratten in doses tot 20 mg/kg/dag (2 maal de humane klinische blootstelling bij volwassen of adolescente patiënten (12 tot 17 jaar) met ITP in een dosis van 75 mg/dag en equivalent aan klinische blootstelling bij mensen bij patiënten met HCV-infectie bij een dosis van 100 mg/dag, blootstellingen gebaseerd op AUC). Bovendien was er geen effect op de embryofoetale ontwikkeling bij konijnen bij doses tot 150 mg/kg/dag, de hoogste geteste dosis (0,3 tot 0,5 maal de humane klinische blootstelling bij ITP-patiënten bij een dosis van 75 mg/dag en bij HCV -geïnfecteerde patiënten in een dosis van 100 mg/dag, blootstellingen op basis van AUC) klinische blootstelling bij mensen bij patiënten met ITP in een dosis van 75 mg/dag en driemaal de klinische blootstelling bij mensen bij patiënten met HCV-infectie bij een dosis van 100 mg/dag, blootstellingen gebaseerd op AUC), werd behandeling met eltrombopag geassocieerd met embryonale letaliteit (verhoogd pre- en post-implantatieverlies), verminderd foetaal lichaamsgewicht en gewicht van de zwangere baarmoeder in het vrouwelijke vruchtbaarheidsonderzoek en een lage incidentie van cervicale ribben en verlaagd foetaal lichaamsgewicht in het onderzoek naar de vrouwelijke vruchtbaarheid embryo-foetale ontwikkelingsstudie Eltrombopag mag alleen tijdens de zwangerschap worden gebruikt als het verwachte voordeel rechtvaardigt het potentiële risico voor de foetus (zie rubriek 4.6). Eltrombopag had geen invloed op de vruchtbaarheid bij mannelijke ratten bij doses tot 40 mg/kg/dag, de hoogste geteste dosis (3 maal de humane klinische blootstelling bij ITP-patiënten bij 75 mg/dag en 2 maal de menselijke klinische blootstelling bij met HCV geïnfecteerde patiënten bij een dosis van 100 mg/dag, blootstellingen gebaseerd op AUC) In de pre- en postnatale ontwikkelingsstudie bij ratten waren er geen nadelige effecten op zwangerschap, bevalling of lactatie van vrouwelijke F0-ratten bij maternale niet-toxische doses (10 en 20 mg/kg/dag) en geen effect op de groei, ontwikkeling, neurologisch gedrag of voortplantingsfunctie van het nageslacht (F1).
Eltrombopag werd gedetecteerd in het plasma van alle nakomelingen van F1-ratten gedurende de volledige 22 uur durende bemonsteringsperiode na toediening van het geneesmiddel aan F0-moeders, wat suggereert dat blootstelling van neonatale ratten aan eltrombopag waarschijnlijk was via borstvoeding.
Opleiding in vitro met eltrombopag wijzen op een mogelijk risico op fototoxiciteit; bij knaagdieren was er echter geen bewijs van cutane fototoxiciteit (10 of 7 keer de menselijke klinische blootstelling bij volwassen of pediatrische ITP-patiënten bij 75 mg / dag en 5 keer de menselijke klinische blootstelling bij patiënten met HCV-infectie bij een dosis van 100 mg / dag, blootstellingen op basis van AUC) of oculaire fototoxiciteit (blootstellingen groter dan 4 maal de klinische blootstelling bij de mens bij volwassen of pediatrische patiënten met ITP in een dosis van 75 mg/dag en 3 maal de klinische blootstelling bij mensen bij met HCV geïnfecteerde patiënten bij een dosis van 100 mg / dag, blootstellingen op basis van AUC). Bovendien toonde een klinisch farmacologisch onderzoek bij 36 proefpersonen geen bewijs dat de fotosensitiviteit was verhoogd na toediening van 75 mg eltrombopag. Dit werd gemeten aan de hand van de vertraagde fototoxische index, maar een potentieel risico op fotoallergie kan niet worden uitgesloten omdat er geen specifieke preklinische onderzoeken kunnen worden uitgevoerd.
Er zijn geen bevindingen bij jonge ratten die wijzen op een groter risico op toxiciteit bij behandeling met eltrombopag bij pediatrische patiënten dan bij volwassenen met ITP.
06.0 FARMACEUTISCHE INFORMATIE
06.1 Hulpstoffen
Revolade 12,5 mg filmomhulde tabletten
Kern van de tablet
Magnesium stearaat
Mannitol (E421)
Microkristallijne cellulose
Povidon (K30)
Natriumzetmeelglycolaat
Tabletcoating
Hypromellose
Macrogol 400
Polysorbaat 80
Titaandioxide (E171)
Revolade 25 mg filmomhulde tabletten
Kern van de tablet
Magnesium stearaat
Mannitol (E421)
Microkristallijne cellulose
Povidon
Natriumzetmeelglycolaat
Tabletcoating
Hypromellose
Macrogol 400
Polysorbaat 80
Titaandioxide (E171)
Revolade 50 mg filmomhulde tabletten
Kern van de tablet
Magnesium stearaat
Mannitol (E421)
Microkristallijne cellulose
Povidon
Natriumzetmeelglycolaat
Tabletcoating
Hypromellose
Rood ijzeroxide (E172)
Geel ijzeroxide (E172)
Macrogol 400
Titaandioxide (E171)
Revolade 75 mg filmomhulde tabletten
Kern van de tablet
Magnesium stearaat
Mannitol (E421)
Microkristallijne cellulose
Povidon
Natriumzetmeelglycolaat
Tabletcoating
Hypromellose
Rood ijzeroxide (E172)
Zwart ijzeroxide (E172)
Macrogol 400
Titaandioxide (E171)
06.2 Incompatibiliteit
Niet relevant.
06.3 Geldigheidsduur
4 jaar.
06.4 Speciale voorzorgsmaatregelen bij bewaren
Voor dit geneesmiddel zijn er geen speciale bewaarcondities.
06.5 Aard van de primaire verpakking en inhoud van de verpakking
Filmomhulde tabletten
Aluminium blisterverpakkingen (PA/Alu/PVC/Alu) in een verpakking met 14 of 28 filmomhulde tabletten en in een multiverpakking met 84 (3 verpakkingen van 28) filmomhulde tabletten.
Mogelijk worden niet alle verpakkingsgrootten in de handel gebracht.
06.6 Instructies voor gebruik en verwerking
Ongebruikte medicijnen en afval afkomstig van dit medicijn moeten worden weggegooid in overeenstemming met de lokale regelgeving.
07.0 HOUDER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Novartis Europharm Limited
Frimley Business Park
Camberley GU16 7SR
VK
08.0 NUMMER VAN DE VERGUNNING VOOR HET IN DE HANDEL BRENGEN
Revolade 12,5 mg filmomhulde tabletten
EU/1/10/612/010
039827100
EU / 1/10/612/011
039827112
EU / 1/10/612/012
039827124
Revolade 25 mg filmomhulde tabletten
EU/1/10/612/001
039827011
EU/1/10/612/002
039827023
EU/1/10/612/003
039827035
Revolade 50 mg filmomhulde tabletten
EU/1/10/612/004
039827047
EU/1/10/612/005
039827050
EU/1/10/612/006
039827062
Revolade 75 mg filmomhulde tabletten
EU/1/10/612/007
039827074
EU/1/10/612/008
039827086
EU / 1/10/612/009
039827098
09.0 DATUM VAN EERSTE VERGUNNING OF VERLENGING VAN DE VERGUNNING
Datum eerste vergunning: 11 maart 2010
Datum van de meest recente verlenging: 15 januari 2015
10.0 DATUM VAN HERZIENING VAN DE TEKST
november 2016







.jpg)